
Competing 1πσ* mediated dynamics in mequinol: O–H versus O–CH3 photodissociation pathways†
Abstract
Deactivation of excited electronic states through coupling to dissociative 1πσ* states in heteroaromatic systems has received considerable attention in recent years, particularly as a mechanism that contributes to the ultraviolet (UV) photostability of numerous aromatic biomolecules and their 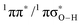 conical intersection (
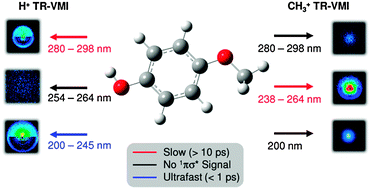
conical intersection (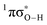 state associated with the O–H bond. In the intermediate wavelength range of 280 ≥ λ ≥ 245 nm,
state associated with the O–H bond. In the intermediate wavelength range of 280 ≥ λ ≥ 245 nm, 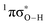 mediated H atom elimination is not observed. In contrast, we find that
mediated H atom elimination is not observed. In contrast, we find that 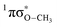 state driven CH3 radical elimination is only observed in the excitation range 264 ≥ λ ≥ 238 nm. Interpretation of these experimental results is guided by: (i) high level complete active space with second order perturbation theory (CASPT2) calculations, which provide 1-D potential energy cuts of the ground and low lying singlet excited electronic states along the O–H and O–CH3 bond coordinates; and (ii) calculated excitation energies using CASPT2 and the equation-of-motion coupled cluster with singles and doubles excitations (EOM-CCSD) formalism. From these comprehensive studies, we find that the dynamics along the O–H coordinate generally mimic H atom elimination previously observed in
state driven CH3 radical elimination is only observed in the excitation range 264 ≥ λ ≥ 238 nm. Interpretation of these experimental results is guided by: (i) high level complete active space with second order perturbation theory (CASPT2) calculations, which provide 1-D potential energy cuts of the ground and low lying singlet excited electronic states along the O–H and O–CH3 bond coordinates; and (ii) calculated excitation energies using CASPT2 and the equation-of-motion coupled cluster with singles and doubles excitations (EOM-CCSD) formalism. From these comprehensive studies, we find that the dynamics along the O–H coordinate generally mimic H atom elimination previously observed in
 Please wait while we load your content...
Please wait while we load your content...

