
Hybrid one-electron/many-electron methods for ionized states of molecular clusters
Abstract
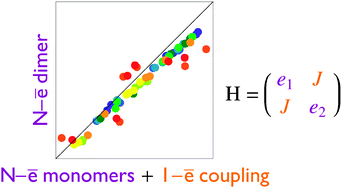
To describe singly-ionized states of molecular clusters we devised an effective Hamiltonian approach that combines (1) accurate monomer ionization potentials from many-electron wave functions with (2) polarization shifts and (3) effective monomer couplings obtained from a simple one-electron approach (the superposition-of-fragment-states (SFS) method [Valeev et al., J. Am. Chem. Soc., 2006, 128, 9882]). The accuracy of the intermolecular coupling parameters evaluated with SFS Hartree–Fock (HF) and Density-Functional-Theory (DFT) variants was evaluated for several weakly-bound dimers and compared against the state-of-the-art equation-of-motion ionization-potential coupled-cluster singles and doubles (EOM-IP-CCSD) data of Krylov and co-workers. The SFS–HF method produces coupling integrals accurate to a few percent, whereas SFS–DFT predictions are substantially worse. A hybrid approach combining SFS–HF couplings and shifts with EOM-IP-CCSD ionization potentials of monomers (denoted as SFS-EOM-IP-CCSD) was applied to ionized states of two conformers of a
- This article is part of the themed collection: Fragment and localized orbital methods in electronic structure theory
 Please wait while we load your content...
Please wait while we load your content...

