
Fragment density functional theory calculation of NMR chemical shifts for proteins with implicit solvation†
Abstract
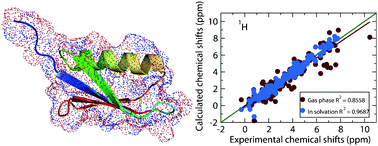
Fragment density functional theory (DFT) calculation of
- This article is part of the themed collection: Fragment and localized orbital methods in electronic structure theory
 Please wait while we load your content...
Please wait while we load your content...

