
Quantum clustering and network analysis of MD simulation trajectories to probe the conformational ensembles of protein–ligand interactions†
Abstract
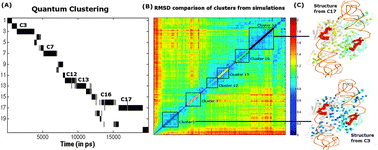
In this article, we present a novel application of a quantum clustering (QC) technique to objectively cluster the conformations, sampled by molecular dynamics simulations performed on different
 Please wait while we load your content...
Please wait while we load your content...