
Hydrogen bond dynamics in heavy water studied with quantum dynamical simulations
Abstract
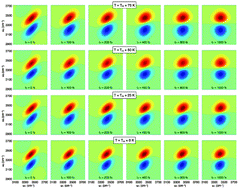
The structure and dynamics of the hydrogen-bond network in heavy water (D2O) is studied as a function of the temperature using quantum dynamical simulations. Our approach combines an ab initio-based representation of the water interactions with an explicit quantum treatment of the molecular motion. A direct connection between the calculated linear and nonlinear
- This article is part of the themed collection: Physics and chemistry of ice and water
 Please wait while we load your content...
Please wait while we load your content...

