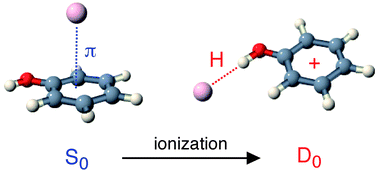
The structures, binding energies, and vibrational and electronic spectra of various isomers of neutral and ionic phenol–Arnclusters with n ≤ 4, PhOH(+)–Arn, are characterized by quantum chemical calculations. The properties in the neutral and ionic ground electronic states (S0, D0) are determined at the M06-2X/aug-cc-pVTZ level, whereas the S1 excited state of the neutral species is investigated at the CC2/aug-cc-pVDZ level. The Ar complexation shifts calculated for the S1 origin and the adiabatic ionisation potential, ΔS1 and ΔIP, sensitively depend on the Ar positions and thus the sequence of filling the first Ar solvation shell. The calculated shifts confirm empirical additivity rules for ΔS1 established recently from experimental spectra and enable thus a firm assignment of various S1 origins to their respective isomers. A similar additivity model is newly developed for ΔIP using the M06-2X data. The isomer assignment is further confirmed by Franck–Condon simulations of the intermolecular vibrational structure of the S1 ← S0 transitions. In neutral PhOH–Arn, dispersion dominates the attraction and π-bonding is more stable than H-bonding. The solvation sequence of the most stable isomers is derived as (10), (11), (30), and (31) for n ≤ 4, where (km) denotes isomers with k and m Ar ligands binding above and below the aromatic plane, respectively. The π interaction is somewhat stronger in the S1 state due to enhanced dispersion forces. Similarly, the H-bond strength increases in S1 due to the enhanced acidity of the OH proton. In the PhOH+–Arn cations, H-bonds are significantly stronger than π-bonds due to additional induction forces. Consequently, one favourable solvation sequence is derived as (H00), (H10), (H20), and (H30) for n ≤ 4, where (Hkm) denotes isomers with one H-bound ligand and k and m π-bonded Ar ligands above and below the aromatic plane, respectively. Another low-energy solvation motif for n = 2 is denoted (11)H and involves nonlinear bifurcated H-bonding to both equivalent Ar atoms in a C2v structure in which the OH group points toward the midpoint of an Ar2 dimer in a T-shaped fashion. This dimer core can also be further solvated by π-bonded ligands leading to the solvation sequence (H00), (11)H, (21)H, and (22) for n ≤ 4. The implications of the ionisation-induced π → H switch in the preferred interaction motif on the isomerisation and fragmentation processes of PhOH(+)–Arn are discussed in the light of the new structural and energetic cluster parameters.

 Please wait while we load your content...
Please wait while we load your content...

