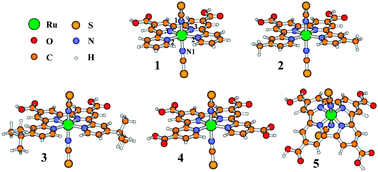
Scalar relativistic density functional theory (DFT) has been used to explore the spectroscopic and redox properties of Ruthenium-type photovoltaic sensitizers, trans-[Ru(RL)(NCS)2] (RL = 4,4′′′-di-R-4′,4′′-bis(carboxylic acid)-2,2′ : 6′,2′′ : 6′′,2′′′-quaterpyridine, R = H (1), Me (2), tBu (3) and COOH (4); RL = 4,4′′′-di-R-4′,4′′-bis(carboxylic acid)-cycloquaterpyridine, R = COOH (5)). The geometries of the molecular ground, univalent cationic and triplet excited states of 1–5 were optimized. In complexes 1–4, the quaterpyridine ligand retains its planarity in the molecular, cationic and excited states, although the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N-Ru angle representing the SCN → Ru coordination approaches 180° in the univalent cationic and triplet excited states. The theoretically designed complex 5 displays a curved cycloquaterpyridine ligand with significantly distorted SCN → Ru coordination. The electron spin density distributions reveal that one electron is removed from the Ru/NCS moieties upon oxidation and the triplet excited state is due to the Ru/NCS → polypyridine charge transfer (MLCT/L'LCT). The experimental absorption spectra were well reproduced by the time-dependent DFT calculations. In the visible region, two MLCT/L'LCT absorption bands were calculated to be at 652 and 506 nm for 3, agreeing with experimental values of 637 and 515 nm, respectively. The replacement of the R- group with -COOH stabilizes the lower-energy unoccupied orbitals of π* character in the quaterpyridine ligand in 4. This results in a large red shift for these two MLCT/L'LCT bands. In contrast, the lower-energy MLCT/L'LCT peak of 5 nearly disappears due to the introduction of cycloquaterpyridine ligand. The higher energy bands in 5 however become broader and more intense. As far as absorption in the visible region is concerned, the theoretically designed 5 may be a very promising sensitizer for DSSC. In addition, the redox potentials of 1–5 were calculated and discussed, in conjunction with photosensitizers such as cis-[Ru(L1)2(X)2] (L1 = 4,4′-bis(carboxylic acid)-2,2′-bipyridine; X = NCS− (6), Cl− (7) and CN− (8)), cis-[Ru(L1′)2(NCS)2] (L1′ = 4,7-bis(carboxylic acid)-1,10-phenanthroline, 9), [NH4][Ru(L2)(NCS)3] (L2 = 4,4′,4′′-tris(carboxylic acid)-2,2′ : 6′,2′′-terpyridine, 10) and [Ru(L2)(NCS)3]− (11).
N-Ru angle representing the SCN → Ru coordination approaches 180° in the univalent cationic and triplet excited states. The theoretically designed complex 5 displays a curved cycloquaterpyridine ligand with significantly distorted SCN → Ru coordination. The electron spin density distributions reveal that one electron is removed from the Ru/NCS moieties upon oxidation and the triplet excited state is due to the Ru/NCS → polypyridine charge transfer (MLCT/L'LCT). The experimental absorption spectra were well reproduced by the time-dependent DFT calculations. In the visible region, two MLCT/L'LCT absorption bands were calculated to be at 652 and 506 nm for 3, agreeing with experimental values of 637 and 515 nm, respectively. The replacement of the R- group with -COOH stabilizes the lower-energy unoccupied orbitals of π* character in the quaterpyridine ligand in 4. This results in a large red shift for these two MLCT/L'LCT bands. In contrast, the lower-energy MLCT/L'LCT peak of 5 nearly disappears due to the introduction of cycloquaterpyridine ligand. The higher energy bands in 5 however become broader and more intense. As far as absorption in the visible region is concerned, the theoretically designed 5 may be a very promising sensitizer for DSSC. In addition, the redox potentials of 1–5 were calculated and discussed, in conjunction with photosensitizers such as cis-[Ru(L1)2(X)2] (L1 = 4,4′-bis(carboxylic acid)-2,2′-bipyridine; X = NCS− (6), Cl− (7) and CN− (8)), cis-[Ru(L1′)2(NCS)2] (L1′ = 4,7-bis(carboxylic acid)-1,10-phenanthroline, 9), [NH4][Ru(L2)(NCS)3] (L2 = 4,4′,4′′-tris(carboxylic acid)-2,2′ : 6′,2′′-terpyridine, 10) and [Ru(L2)(NCS)3]− (11).

 Please wait while we load your content...
Please wait while we load your content...

