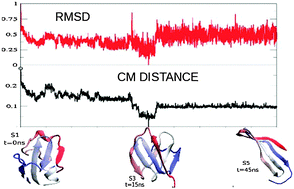
An important step in the computer simulation of the dynamics of biomolecules is the comparison of structures in a trajectory by exploiting a measure of distance. This allows distinguishing structures which are geometrically similar from those which are different. By analyzing microseconds-long all-atom molecular dynamics simulations of a polypeptide, we find that a distance based on backbone dihedral angles performs very well in distinguishing structures that are kinetically correlated from those that are not, while the widely used Cα root mean square distance performs more poorly. The root mean square difference between contact matrices turns out instead to be the metric providing the highest clustering coefficient, namely, according to this similarity measure, the neighbors of a structure are also, on average, neighbors among themselves. We also propose a combined distance measure which, for the system considered here, performs well both for distinguishing structures which are distant in time and for giving a consistent cluster analysis.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


