
Can P–H σ-bond complexes be prepared? A computational study by DFT and AIM methods†
Abstract
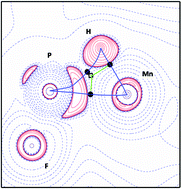
A series of complexes of general formula [CpMn(CO)2(η2-HPR1R2·BCl3)] has been studied by DFT calculations and topological analyses of the charge density thus derived. The 21 complexes included in this study exhibit closely similar Mn–H–P geometries, in spite of a wide range of substituents (R1; R2) at the phosphorus atom. Topological analysis of the electron density suggests that these are genuine σ-bond complexes, albeit at a later stage on the oxidative
 Please wait while we load your content...
Please wait while we load your content...

