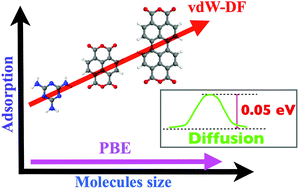
The self-assembly of flat organic molecules on metal surfaces is controlled, apart from the kinetic factors, by the interplay between the molecule–molecule and molecule–surface interactions. These are typically calculated using standard density functional theory within the generalized gradient approximation, which significantly underestimates nonlocal correlations, i.e. van der Waals (vdW) contributions, and thus affects interactions between molecules and the metal surface in the junction. In this paper we address this question systematically for the Au(111) surface and a number of popular flat organic molecules which form directional hydrogen bonds with each other. This is done using the recently developed first-principles vdW-DF method which takes into account the nonlocal nature of electron correlation [M. Dion et al., Phys. Rev. Lett. 2004, 92, 246401]. We report here a systematic study of such systems involving completely self-consistent vdW-DF calculations with full geometry relaxation. We find that the hydrogen bonding between the molecules is only insignificantly affected by the vdW contribution, both in the gas phase and on the gold surface. However, the adsorption energies of these molecules on the surface increase dramatically as compared with the ordinary density functional (within the generalized gradient approximation, GGA) calculations, in agreement with available experimental data and previous calculations performed within approximate or semiempirical models, and this is entirely due to the vdW contribution which provides the main binding mechanism. We also stress the importance of self-consistency in calculating the binding energy by the vdW-DF method since the results of non-self-consistent calculations in some cases may be off by up to 20%. Our calculations still support the usually made assumption of the molecule–surface interaction changing little laterally suggesting that single molecules and their small clusters should be quite mobile at room temperature on the surface. These findings support a gas-phase modeling for some flat metal surfaces, such as Au(111), and flat molecules, at least as a first approximation.

 Please wait while we load your content...
Please wait while we load your content...

