Under the usual assumption of noninteracting v-representability, density-functional theory (DFT) together with time-dependent DFT (TDDFT) provide a formally exact single-reference method suitable for the theoretical description of the electronic excited-states of large molecules, and hence for the description of excited-state potential energy surfaces important for photochemistry. The quality of this single-reference description is limited in practice by the need to use approximate exchange–correlation functionals. In particular it is far from clear how well approximations used in contemporary practical TDDFT calculations can describe funnel regions such as avoided crossings and conical intersections. These regions typically involve biradical-like structures associated with bond breaking and conventional wisdom would seem to suggest the need to introduce explicit double excitation character to describe these structures. Although this is lacking in ordinary spin-preserving (SP) TDDFT, it is present to some extent in spin-flip (SF) TDDFT. We report our tests of Wang–Ziegler noncollinear SF-TDDFT within the Tamm–Dancoff approximation for describing the avoided crossing in the C2v CC ring-opening reaction of oxirane and for describing the conical intersection relevant for the more physical asymmetric CO ring-opening reaction of oxirane. Comparisons are made with complete active space self-consistent field and quantum Monte Carlo benchmark results from two previous papers on the subject [J. Chem. Phys., 2007, 127, 164111; ibid129, 2008, 124108]. While the avoided crossing in the C2v pathway is found to be reasonably well described, the method was found to be only partially successful for the conical intersection (CX) associated with the physically more important asymmetric pathway. The origin of the difficulties preventing the noncollinear SF-TDDFT method from giving a completely satisfactory description of the CX was traced back to the inability of SF-TDDFT based upon a single triplet reference state to correlate all potentially relevant configurations involving not just two but three nearly degenerate orbitals (n, σCO, and 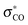 ). This article is also the first report of our implementation of SF-TDDFT within the DEMON2K program.
). This article is also the first report of our implementation of SF-TDDFT within the DEMON2K program.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


