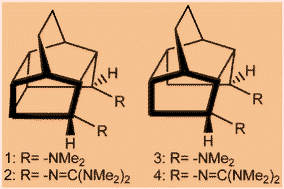
According to DFT calculations, pentacyclo[6.4.0.02,7.03,11.06,10]dodecane (PCD) derivatives have been predicted as organic super bases. The new molecular framework (PCD) is versatile in terms of anchoring different functional groups to achieve high basicities in both gas and solvent phase. Density functional quantum chemical calculations at the B3LYP/6-311+G**//B3LYP/6-31G* level have been performed to calculate the proton affinities of pentacyclo[6.4.0.02,7.03,11.06,10]dodecane (PCD) derivatives and substituted pentacyclo[5.4.0.02,6.03,10.05,9]undecane (PCU) derivatives. The calculated results suggest that these polycyclic cage units can act as organic superbases when anchored with different functional groups. The flexibility in these polycyclic rings allowed the attainment of better hydrogen bonding compared to the prototype 1,8-bis(dimethyl-amino)naphthalene and 1,8-bis(diguanidino)naphthalene. The predicted higher basicities appeared to be due to the interplay of steric repulsions and the strength of hydrogen bonding in these cases. Intramolecular and intermolecular proton transfer barriers have been calculated for these amines and imines.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


