
Towards a gauge invariant method for molecular chiroptical properties in TDDFT
Abstract
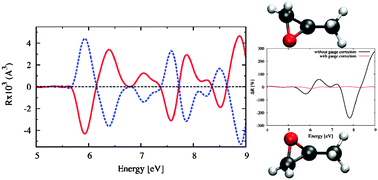
We present an efficient scheme to calculate the chiroptical response of molecular systems within time dependent density functional theory using either a real-time propagation or a frequency-dependent Sternheimer method. The scheme avoids the commonly used sum over empty orbitals and has a very favorable scaling with system size. Moreover, the method is general and can be easily implemented. In the present work, we implemented it using a real-space pseudo-potential representation of the wave-functions and Hamiltonian. The specific use of non-local pseudo-potentials implies that a gauge correction term in the angular momentum operator must be included to ensure that the total scheme is fully gauge invariant. Applications to small organic chiral molecules are shown and discussed, addressing some deficiencies of present exchange–correlation functionals to describe the absolute position of the excitations. However, the shape or sign of the dichroism spectra comes out in excellent agreement with available experiments.
- This article is part of the themed collection: Time-dependent density-functional theory
 Please wait while we load your content...
Please wait while we load your content...

