
First-principles semiclassical initial value representation molecular dynamics
Abstract
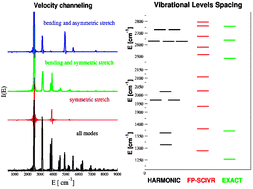
In this work, we explore the use of the semiclassical initial value representation (SC-IVR) method with first-principles electronic structure approaches to carry out classical molecular dynamics. The proposed approach can extract the vibrational power
- This article is part of the themed collection: Celebrating the centenary of the Italian Chemical Society
 Please wait while we load your content...
Please wait while we load your content...

