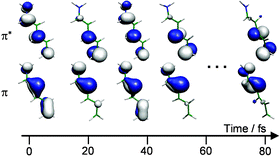
Multireference ab initio dynamics simulations have become available as a tool for the investigation of photochemical processes, mainly for those related to nonadiabatic phenomena taking place in the sub-picosecond time scale. For organic molecules, these phenomena are in many cases deeply dependent on the relaxation of the photoexcited π-system. We review the latest contributions of our group to this subject and report new results for systems studied previously, grouping them in single π bonds, chains and aromatic rings. The dynamics of ethylene and substituted ethylenes is discussed mainly in connection to the competition between the two available relaxation paths in the excited states and their relation to the conical intersections in large systems. The trans–cis and the cis–trans dynamics of the pentadieniminium cation is investigated as well. Finally, we discuss the photodynamics of aminopyrimidine starting in the S1 and S2 states and the conclusions, which can be drawn from this for the interpretation of the adenine dynamics.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


