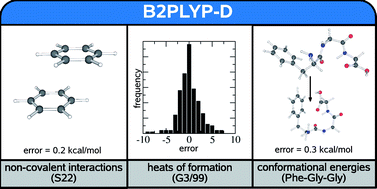
The objective of this work is the further systematic improvement of the accuracy of Double-Hybrid Density Functionals (DHDF) that add non-local electron correlation effects to a standard hybrid functional by second-order perturbation theory (S. Grimme, J. Chem. Phys., 2006, 124, 034108). The only known shortcoming of these generally highly accurate functionals is an underestimation of the long-range dispersion (van der Waals) interactions. To correct this deficiency, we add a previously developed empirical dispersion term (DFT-D) to the energy expression but leave the electronic part of the functional untouched. Results are presented for the S22 set of non-covalent interaction energies, the G3/99 set of heat of formations and conformational energies of a phenylalanyl–glycyl–glycine peptide model. We furthermore propose seven hydrocarbon reactions with strong intramolecular dispersion contributions as a benchmark set for newly developed density functionals. In general, the proposed composite approach is for many chemically relevant properties of similar quality as high-level coupled-cluster treatments. A significant increase of the accuracy for non-covalent interactions is obtained and the corrected B2PLYP DHDF provides one of the lowest ever obtained Mean Absolute Deviations (MAD) for the S22 set (0.2–0.3 kcal mol−1). Unprecedented high accuracy is also obtained for the relative energies of peptide conformations that turn out to be very difficult. The significant improvements found for the G3/99 set (reduction of the MAD from 2.4 to 1.7 kcal mol−1) underline the importance of intramolecular dispersion effects in large molecules. In all tested cases the results from the standard B3LYP approach are also significantly improved, and we recommend the general use of dispersion corrections in DFT treatments.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


