
The length, strength and polarity of metal–carbon bonds: dialkylzinc compounds studied by density functional theory calculations, gas electron diffraction and photoelectron spectroscopy†
Abstract
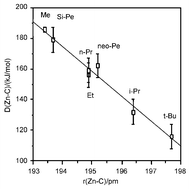
The molecular structures and thermodynamic functions of seven dialkyl zinc compounds, R2Zn, R = Me, Et, i-Pr, t-Bu, n-Pr, neopentyl and the silaneopentyl Me3SiCH2, of the parent hydrocarbons RH and of the radicals R have been determined by density functional theory calculations at the B3LYP/SDD level. The molecular structures of the i-Pr, t-Bu, neo-Pe and Me3SiCH2 derivatives have been determined by gas electron diffraction. Me2Zn, Et2Zn, t-Bu2Zn and neo-Pe2Zn have been studied by photoelectron spectroscopy and the ionisation energies calculated. Both experimental and calculated Zn-C bond distances were found to increase in the order
Calculated mean bond rupture enthalpies indicate that the strength of the Zn–C bonds decrease in the same order, viz.
Both bond lengths and bond strengths were found to be strongly correlated with the inductive Taft constant I, indicating that the bond strength increases and the bond length decreases with increasing electron withdrawing power of the alkyl group. Evidence from the literature indicates that bond strengths and bond lengths in homoleptic alkyl derivatives of the main group metals in Groups 12, 13 and 14 and of the transition elements in Group 4 vary in the same manner.
 Please wait while we load your content...
Please wait while we load your content...

