
Ab initio predictions of ferroelectric ternary fluorides with the LiNbO3 structure†
Abstract
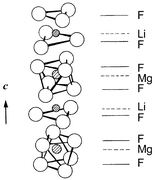
First-principles periodic density-functional theory calculations suggest ternary fluorides LiMgF3, NaCaF3 and LiNiF3 should adopt the ferroelectric LiNbO3 structure at low temperatures; LiMgF3 and LiNiF3 are predicted to have negative enthalpies of formation from the binary fluorides.
 Please wait while we load your content...
Please wait while we load your content...

