
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed regioselective and stereo-invertive ring-opening borylation of 2-arylaziridines with bis(pinacolato)diboron: experimental and computational studies†
Youhei
Takeda
 *a,
Akinobu
Kuroda
a,
W. M. C.
Sameera
b,
Keiji
Morokuma
*b and
Satoshi
Minakata
*a
*a,
Akinobu
Kuroda
a,
W. M. C.
Sameera
b,
Keiji
Morokuma
*b and
Satoshi
Minakata
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan. E-mail: takeda@chem.eng.osaka-u.ac.jp; minakata@chem.eng.osaka-u.ac.jp
bFukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishihiraki-cho 34-4, Sakyo-ku, Kyoto 606-8103, Japan. E-mail: morokuma.keiji.3a@kyoto-u.ac.jp
First published on 9th June 2016
Abstract
A palladium catalyzed regioselective borylative ring opening reaction of 2-arylaziridines to give β-amino-β-arylethylborates was developed. The reaction reported herein represents the first example of ring-opening borylation of non-vinylic aziridines and direct borylative C(sp3)–N bond cleavage of neutral organic substrates. NMR studies and density functional theory (DFT) calculations suggested that the active intermediate for the reaction is a PdL2 complex [L = P(t-Bu)2Me]. The multi-component artificial force-induced reaction method (MC-AFIR) located the transition states for the regioselectivity-determining aziridine ring opening that proceeds in an SN2 fashion, and explained the selectivity of the reaction. The full catalytic cycle consists of a selectivity-determining aziridine ring opening (oxidative addition), a proton transfer, phosphine ligand dissociation from the catalyst, boron–boron bond cleavage, and reductive elimination. Water is important to the drive the transmetalation step. The calculated overall mechanism and selectivity are consistent with the experimental results.
Introduction
Aziridines, three-membered strained azaheterocycles, serve as versatile building blocks in modern organic synthesis.1 Regioselective ring opening of aziridines with nucleophiles, which is driven by release of its ring strain, is one of the most useful transformations of aziridines into ubiquitous β-amino-functionalized motifs.2 Aziridines undergo oxidative addition to low-valent late transition metal complexes at the terminal C–N bond in an SN2 fashion, generating the corresponding oxidative adducts, azametallacyclobutanes.3–5 In light of this reactivity, diverse transition metal-catalyzed regio-, stereo-, and/or chemoselective transformations of aziridines are feasible,6–9 where the oxidative addition is smoothly coupled with subsequent elementary processes, such as transmetalation, migratory insertion, and reductive elimination; regioselective Rh- and Co-catalyzed carbonylative ring expansion of aziridines, pioneered by Alper and co-workers, represent the embodiment of this idea.6Over the past few years, numerous advances have been made in transition metal-catalyzed ring-opening cross coupling of aziridines, where aziridines can be utilized as a non-classical alkyl electrophilic partner [eqn (1)].10,11 In 2012, Doyle reported the first Ni-catalyzed regioselective Negishi alkylation of 2-aryl-N-tosylaziridines that leads to branch-type products via the regioselective cleavage of a benzylic C–N bond.10a Since this report, several groups including us11b have developed Ni-10 and Pd-catalyzed11 regioselective ring-opening cross coupling reactions of aziridines to form C(sp3)–C(sp2)/C(sp3) bonds, with the regioselectivity seemingly governed by the aziridine substrate rather than the catalyst [eqn (1)]. Despite the above-mentioned successes in C–C cross couplings, other catalytic ring-opening C–E (E ≠ C) bond forming reactions of aziridines based on a cross-coupling mechanism are also unexploited.12
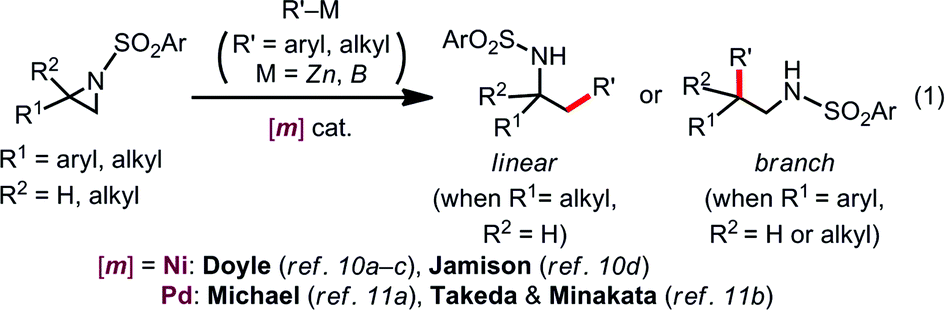
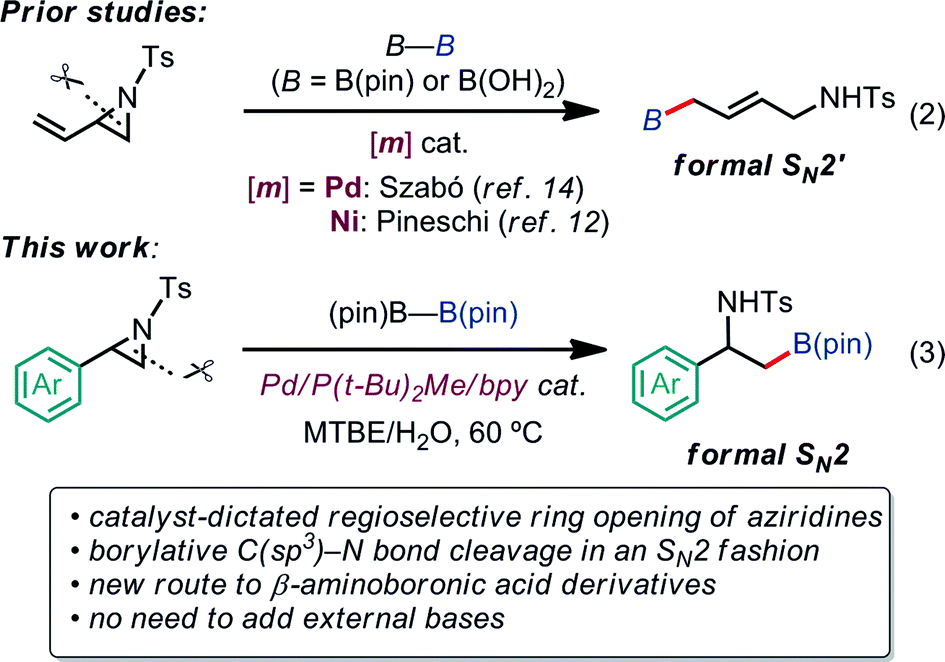
Our latest findings11b led us to seek new catalytic ring-opening C–E coupling systems of aziridines. Moreover, we became intrigued with the C–B coupling of aziridines to give β-amino-functionalized alkylboronates; alkylboronic acid derivatives serve not only as useful building blocks in organic synthesis, but are also biologically interesting motifs in medicinal chemistry.13 Only a few precedents in transition-metal-catalyzed borylative substitution of vinylaziridines via C(sp3)–N bond cleavage have been reported. Szabó and Pineschi disclosed a Pd(II) pincer complex14 and a Ni(0)/BINAP12 system as catalysts for the borylative ring opening of vinylaziridines with diboron reagents via the cleavage of an allylic C–N bond to give γ-amino alkylboronic acid derivatives (formal SN2′-type reaction),15 respectively [eqn (2)]. Nevertheless, to the best of our knowledge, the catalytic direct displacement of the C(sp3)–N bond of aziridines with a C(sp3)–B bond on the same carbon (formal SN2-type reaction) has never been described to date.
We herein report a regioselective ring-opening C–B cross-coupling reaction of 2-arylaziridines that is realized using a Pd/P(t-Bu)2Me/bpy catalytic system to give β-amino-alkylboronates [eqn (3)], which can serve as versatile building blocks to synthesize amino-functionalized compounds and biologically relevant β-amino acid surrogates.16,17 Notably, our reaction features the opposite regioselectivity in C–N bond cleavage (at the 3-position) to those previously reported for C–C cross couplings using the same type of aziridine substrate (at the 2-position).10a,c,11b Also, our C–B coupling system represents the first example of a formal SN2-type borylative C(sp3)–N bond cleavage of neutral compounds.18 It should be noted that the present C–B coupling proceeds smoothly under neutral conditions as in the cases of borylative substitution of allyl carboxylates19 and carbonates.19a,20 In these cases, the leaving group-derived oxy anions (RCO2− and RO−) serve as the internal bases, while the metal catalyzed borylative substitution of alkyl halides usually requires the addition of stoichiometric external base or activator to promote transmetalation.21 In addition to experimental elaboration, we have performed theoretical calculations, applying density functional theory (DFT) and the multi-component artificial force-induced reaction (MC-AFIR) method to determine the mechanism of the reaction and explain the origin of the selectivity, and to clarify why external base is not required in this reaction system.
Results and discussion
Experimental part
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Difference from the “standard conditions” | Yielda (%) | Recovery of 1aa (%) | |||
| 3a | 4 | 5 | 6 | |||
| a Determined with GC or 1H NMR. b 1 mol% of Cp(allyl)Pd and 2 mol% of ligand were used. c 1 mol% of Cp(allyl)Pd and 0.5 mol% of ligand were used. | ||||||
| 1 | None | 81 | 0 | 0 | 13 | 0 |
| 2 | Cp(allyl)Pd (1 mol%)/P(t-Bu)2Me (2 mol%) | 74 | 0 | 11 | 12 | 3 |
| 3 | Cp(allyl)Pd (1 mol%)/P(t-Bu)2Me (3 mol%) | 45 | 0 | 10 | 14 | 0 |
| 4b | Without bpy | 52 | 0 | 14 | 25 | 0 |
| 5b |
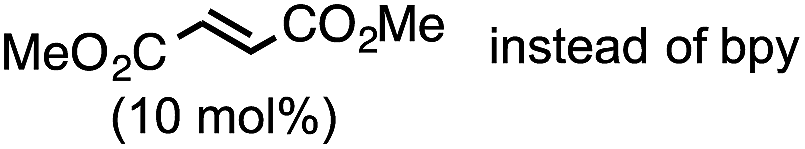
|
0 | 0 | 10 | 0 | 90 |
| 6b |

|
Trace | 0 | 3 | 0 | 96 |
| 7b |
|
64 | 0 | 8 | 16 | 0 |
| 8 | Pd2(dba)3/SIPr was used as catalyst | 0 | 5 | 37 | 0 | 48 |
| 9 | Pd2(dba)3/PCy3 was used as catalyst | 5 | 10 | 0 | 0 | 85 |
| 10 | Pd[P(t-Bu)3]2 was used as catalyst | 0 | 0 | 0 | 0 | 97 |
| 11c | Without H2O | 0 | 0 | 0 | 7 | 84 |
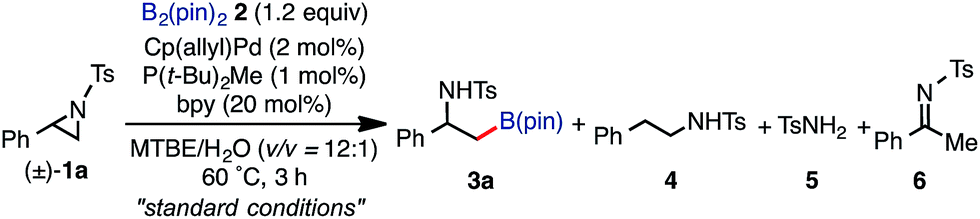
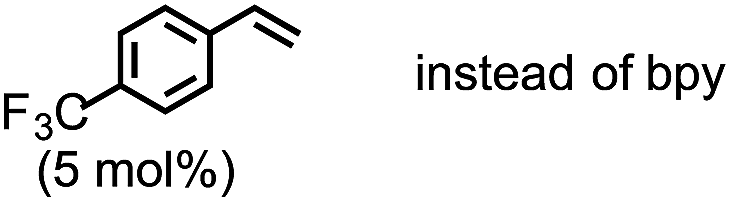
The highest yield (81%, isolated yield: 71%) of borylated product 3a was obtained when 1a was treated with 1.2 equiv. of B2(pin)2 at 60 °C in the presence of catalytic amounts of Cp(allyl)Pd (2 mol%), P(t-Bu)2Me23 (1 mol%), and 2,2′-bipyridine (bpy, 20 mol%) in a mixed solvent of methyl-tert-butylether (MTBE) and H2O (entry 1, “standard conditions”). Most importantly, the regioselectivity of this ring opening and C–B coupling was opposite to those observed with the C–C coupling of the same substrate,10a,c,11b implying that the oxidative addition occurred at the C–N bond on the terminal carbon (the 3-position of the aziridine). The regioisomer of 3a was not detected in the crude 1H NMR spectra, indicating that the regioselectivity in the ring-opening of 1a should be almost perfect. In fact, the regioselective oxidative addition dictated by the interactions between the substrate and the Pd(0) catalyst was supported by theoretical calculations (vide infra). The L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio [L = P(t-Bu)2Me] was found to have a significant impact on the product distribution (entries 2 and 3). As the L:Pd ratio was increased from 0.5 (entry 1) to 2 to 3, the yields of 3a decreased to 74% and 45%, respectively (entries 2 and 3). This irregular ratio of L:Pd implies that there might be a complex equilibrium of PdLn species generated in situ, some of which are specifically active in the catalytic cycle. In fact, this speculation was partly supported with 31P NMR experiments and theoretical calculations (vide infra). Addition of electron deficient unsaturated compounds, which can coordinate to metal complexes, is an effective strategy to suppress undesired β-hydride elimination or to accelerate reductive elimination, thereby often leading to better results in alkyl cross coupling reactions.24 Indeed, the effect of adding bpy was significantly effective for suppressing the formation of byproduct 6 as well as 5, which was presumably derived from the hydrolysis of 6 with H2O (entries 4–7). The effect of the ligands was also significant (entries 8–10). N-heterocyclic carbene (NHC) ligands, SIPr for instance,11b promoted the consumption of 1a and produced undesired products 4 and 5 (entry 8). The results obtained from sterically demanding trialkylphosphine/Pd catalysts (entries 9 and 10) suggest a sluggish oxidative addition step, which is in good agreement with Wolfe's report.9 Furthermore, as mentioned in the introduction section, the ring opening borylation disclosed herein does not require the addition of any external base, which is usually used to activate the transmetalation step for the borylative substitution of alkyl halides. In connection with this, addition of water is crucial. Moreover, in the absence of H2O, no conversion of 1a was observed (entry 11), while at least 1 equiv. of H2O allowed the borylation (Table S11 in the ESI†). Theoretical calculations implied that H2O serves as a proton source (H+) as well as a source of internal base (Pd–OH) to promote the four-membered transmetalation between B2(pin)2 and Pd (vide infra). To investigate the fate of the other B(pin) moiety of B2(pin)2 in the reaction, the 11B NMR spectrum of the crude product was acquired in benzene-d6. The only peak other than the remaining B2(pin)2 was detected at δ = 22.6 ppm as a slightly broad singlet. The comparison with the reported 11B resonances of (pin)B–OH (δ = 22.5 ppm in benzene-d6)25a and (pin)B–O–B(pin) (δ = 21.6 ppm in benzene-d6)25b would suggest that one of the B(pin) moieties of the starting diboron reagent was converted to (pin)B–OH during the reaction.
:Pd ratio [L = P(t-Bu)2Me] was found to have a significant impact on the product distribution (entries 2 and 3). As the L:Pd ratio was increased from 0.5 (entry 1) to 2 to 3, the yields of 3a decreased to 74% and 45%, respectively (entries 2 and 3). This irregular ratio of L:Pd implies that there might be a complex equilibrium of PdLn species generated in situ, some of which are specifically active in the catalytic cycle. In fact, this speculation was partly supported with 31P NMR experiments and theoretical calculations (vide infra). Addition of electron deficient unsaturated compounds, which can coordinate to metal complexes, is an effective strategy to suppress undesired β-hydride elimination or to accelerate reductive elimination, thereby often leading to better results in alkyl cross coupling reactions.24 Indeed, the effect of adding bpy was significantly effective for suppressing the formation of byproduct 6 as well as 5, which was presumably derived from the hydrolysis of 6 with H2O (entries 4–7). The effect of the ligands was also significant (entries 8–10). N-heterocyclic carbene (NHC) ligands, SIPr for instance,11b promoted the consumption of 1a and produced undesired products 4 and 5 (entry 8). The results obtained from sterically demanding trialkylphosphine/Pd catalysts (entries 9 and 10) suggest a sluggish oxidative addition step, which is in good agreement with Wolfe's report.9 Furthermore, as mentioned in the introduction section, the ring opening borylation disclosed herein does not require the addition of any external base, which is usually used to activate the transmetalation step for the borylative substitution of alkyl halides. In connection with this, addition of water is crucial. Moreover, in the absence of H2O, no conversion of 1a was observed (entry 11), while at least 1 equiv. of H2O allowed the borylation (Table S11 in the ESI†). Theoretical calculations implied that H2O serves as a proton source (H+) as well as a source of internal base (Pd–OH) to promote the four-membered transmetalation between B2(pin)2 and Pd (vide infra). To investigate the fate of the other B(pin) moiety of B2(pin)2 in the reaction, the 11B NMR spectrum of the crude product was acquired in benzene-d6. The only peak other than the remaining B2(pin)2 was detected at δ = 22.6 ppm as a slightly broad singlet. The comparison with the reported 11B resonances of (pin)B–OH (δ = 22.5 ppm in benzene-d6)25a and (pin)B–O–B(pin) (δ = 21.6 ppm in benzene-d6)25b would suggest that one of the B(pin) moieties of the starting diboron reagent was converted to (pin)B–OH during the reaction.
|
|
|---|
|
a Reaction conditions: 1 (0.50 mmol), 2 (0.60 mmol), Cp(allyl)Pd (10 μmol), P(t-Bu)2Me (5 μmol), bpy (10 μmol) in MTBE/H2O (1.5 mL, v/v 12:1) at 60 °C under N2 atmosphere for 3 h.
b The values outside and inside parentheses indicate 1H NMR and isolated yields, respectively.
c The reaction was conducted at 80 °C.
|
|
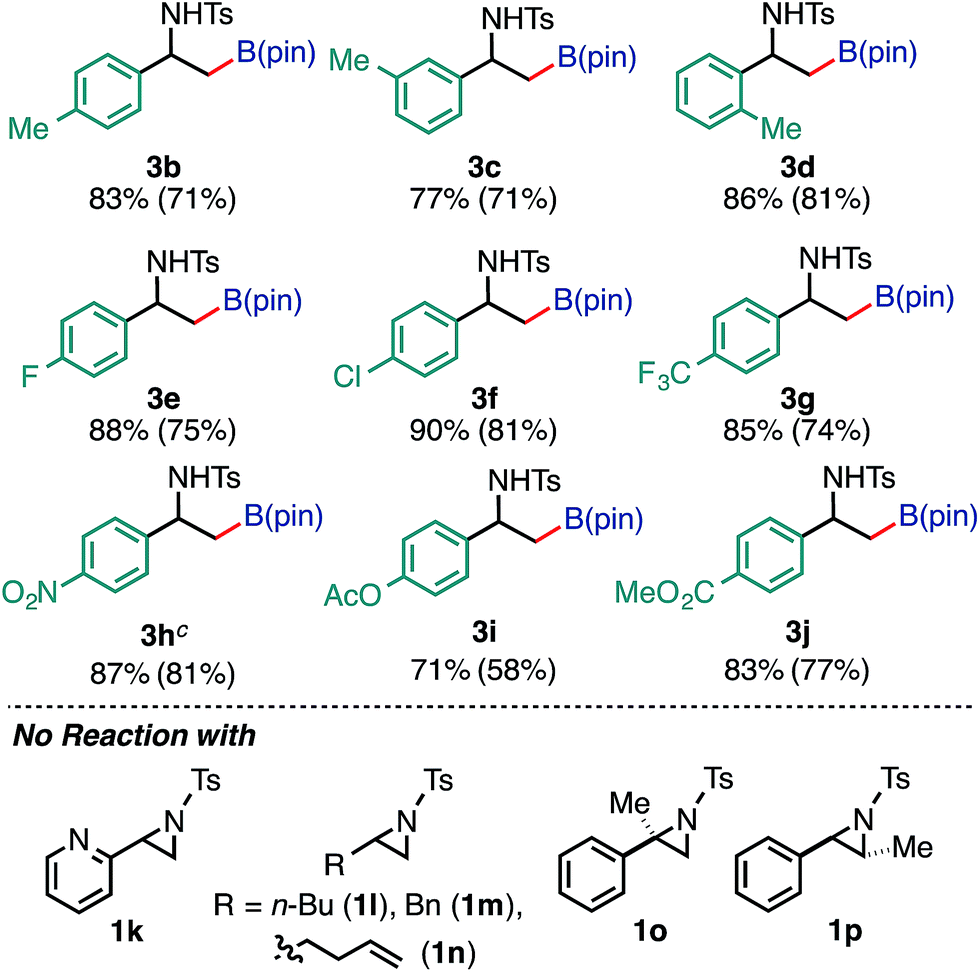
Furthermore, the borylation was scalable, and 3a was prepared on a gram scale (2.89 g, 72% yield) from 10 mmol of 1a (pS17 of the ESI†). With respect to the limitation of substrate, 2-pyridyl- (1k) and 2-alkylaziridines (1l, 1m, and 1n) were not consumed under the reaction conditions. Furthermore, this reaction is very sensitive to the steric hindrance around the 3-positioned carbon, where oxidative addition to Pd(0) has occurred. For example, 2-methyl-2-phenylaziridine (1o) and trans-2-phenyl-3-methylaziridine (1p) were not consumed at all under the standard conditions. As for the scope of the N-functional groups of aziridines, p-MeOC6H4SO2 and t-BuSO2-substituted 2-phenylaziridines smoothly underwent borylation in a regioselective manner (at the 3-position) to produce the corresponding β-amino-β-phenylethylboronates 3r (65%) and 3s (52%), respectively. On the other hand, N-p-nosyl-2-phenylaziridine did not give borylated product (Table S12 in the ESI†). The use of an N-acylated aziridine, N-Boc-2-phenylaziridine for instance, did not give borylated products at a noticeable level, highlighting the efficacy of the N-tosyl group for the borylation. Regarding the diboron reagents, the use of bis(neopentyl glycolato)diboron gave the corresponding aminoboronate product (3a′) in 39% yield, while the borylation using bis(hexylene glycolato)diboron produced the corresponding borylated products in a low yield as an inseparable diastereomeric mixture, presumably due to the existence of two chiral centers on the benzylic and glycolate carbons (Table S13 in the ESI†).
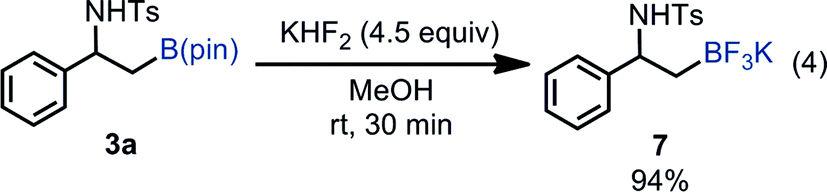
Borylated product 3a was successfully transformed into β-amino-β-aryl-substituted alkyltrifluoroborate 7 in excellent yield, which can serve as a versatile building block to synthesize β-amino-β-arylethanes through Pd-catalyzed cross coupling reactions [eqn (4)].26
In order to understand the stereochemical outcome of the secondary stereogenic center of the borylated product and to demonstrate the utility of the alkylboronate, an enantiopure aziridine (R)-1a (99% ee from chiral HPLC analysis) was subjected to the standard reaction conditions (Scheme 1). HPLC analysis of the borylated product 3a revealed that the stereochemical information was completely retained (99% ee, S) through the reaction. This result excludes the possibility of the reaction pathway of oxidative addition/β-hydride elimination/re-insertion of [Pd–H] species leading to 3a, although it cannot perfectly exclude such a pathway.27 To demonstrate the synthetic utility of the enantiopure β-amino-alkylboronate (S)-3a, the asymmetric synthesis of 3-phenyl-1,2,3,4-tetrahydroisoquinoline derivative was conducted (Scheme 1). The tetrahydroisoquinoline skeleton is a ubiquitous motif in alkaloid natural products and constitutes a biologically important azaheterocycle.28 Enantiopure alkylboronate (S)-3a was subjected to the Pd-catalyzed Suzuki–Miyaura cross coupling29 with chlorobenzene to give the optically active 1,2-diphenylamine derivative (S)-8 in 69% yield, while keeping the enantiopurity intact (99% ee, chiral HPLC). The following Pictet–Spengler reaction30 successfully gave the enantiopure tetrahydroisoquinoline product (S)-9 in excellent yield.
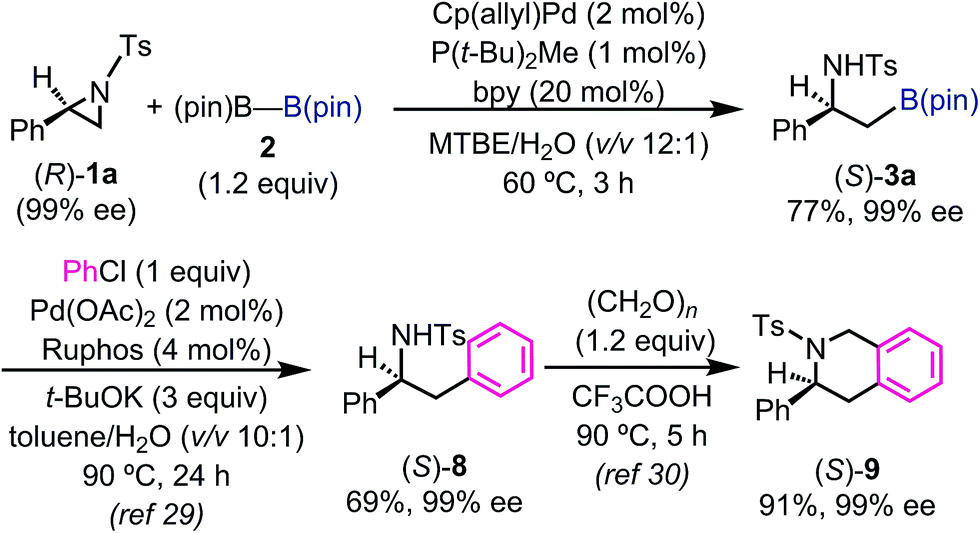 | ||
| Scheme 1 Preparation of enantiopure β-aminoboronate and its synthetic application. | ||
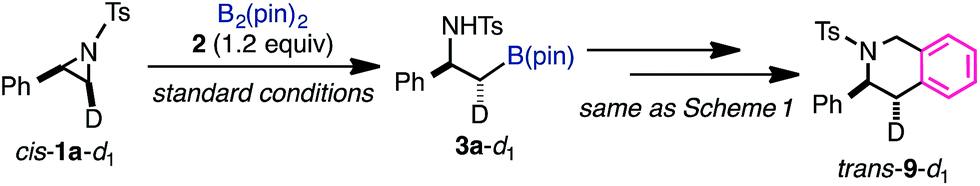 | ||
| Scheme 2 Borylation of deuterated aziridine. | ||
To identify the Pd species generated by mixing P(t-Bu)2Me and Cp(allyl)Pd, we monitored the 31P{1H} NMR spectra of these mixtures in THF-d8 at 60 °C (Fig. 1). Upon addition of 2 equiv. of Cp(allyl)Pd to L (i.e., L:Pd = 0.5), the sharp resonance corresponding to free L at δ 15.3 ppm (Fig. 1a, lit. δ 12.5 ppm at room temperature in toluene-d8),31 completely disappeared and three new peaks appeared at δ 37.0 (sharp, strong), 44.7 (broad, weak), and 62.0 (broad, weak) ppm (Fig. 1b). According to the data reported by the Baird group,32 these peaks are assignable to a dinuclear Pd(I) complex [Pd2L2(μ-Cp)(μ-allyl)] (lit. δ 35.4 ppm at 65 °C in toluene-d8), a bisphosphine Pd(0) complex PdL2 (lit. δ 41.0 ppm at 65 °C in toluene-d8), and an [(η5-Cp)Pd(η1-allyl)L] species ([(η5-Cp)Pd(η1-PhC3H4)L] lit. δ 61.7 ppm at room temperature in toluene-d8), respectively, as illustrated in Fig. 1. After stirring the mixture at 60 °C for 1 h, the broadened weak peak at δ 62.0 ppm corresponding to [(η5-Cp)Pd(η1-allyl)L] disappeared, and the intensity of the peak at δ 44.7 ppm (PdL2) increased as well (the integration ratio of [Pd2L2(μ-Cp)(μ-allyl)]:PdL2 = 3:2, Fig. 1c). These peaks were not changed when bpy (10 equiv.) was added (Fig. S1b in the ESI†). This result would indicate that bpy does not coordinate to these Pd species, which was also supported by our computational results (vide infra). Moreover, upon addition of another equivalent of L (i.e., L:Pd = 1), the strongest sharp peak at δ 37.0 ppm (dinuclear Pd complex) disappeared, and a broad peak around δ 42.6 ppm appeared (Fig. 1d). Furthermore, addition of one more equivalent of L (L:Pd = 2) gave a significantly broadened peak (δ ∼ 30 ppm, Fig. S1a in the ESI†), indicating significant exchange among several PdLn species. Our observations could indicate that PdL2 undergoes rapid exchange with PdLn in THF-d8 when the L:Pd ratio is greater than 0.5. Therefore, the same would be the case with the real reaction system.
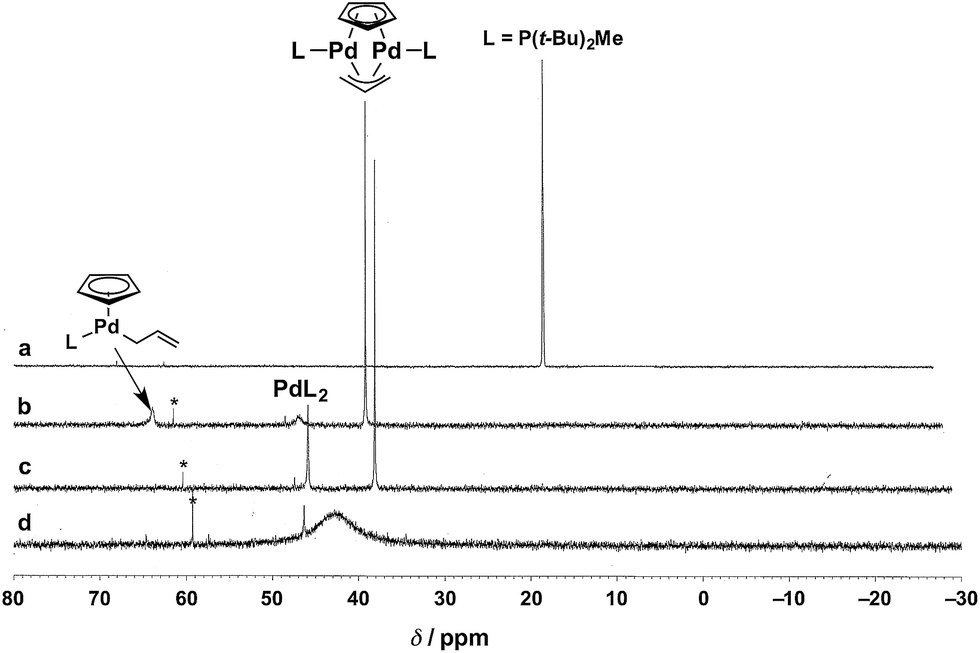 | ||
| Fig. 1 Stacked 31P{1H} NMR spectra acquired at 60 °C in THF-d8. (a) Free L [L = P(t-Bu)2Me]; (b, c) the reaction of Cp(allyl)Pd with 0.5 equiv. of L ((b) upon mixing, (c) 1 h); (d) the reaction of Cp(allyl)Pd with 1 equiv. of L after 1 h. The peaks with asterisks are attributed to phosphine oxide. | ||
Based on the NMR experiments, [Pd2L2(μ-Cp)(μ-allyl)] or PdL2 would be the catalytically active Pd species (i.e., the starting point of the catalytic cycle). To determine which is the active species in the catalytic reaction, both complexes were generated separately as the sole components according to Baird's conditions (i.e., 1:1 and 2:1 mixtures of L and Cp(allyl)Pd were mixed in toluene-d8 and stirred at 77 °C for 1 h),32 and the resulting Pd complexes were used as a catalyst in the borylation reaction in toluene (Schemes 3). As a result, while the dinuclear Pd(I) complex was found to be totally inert for the borylation reaction, the bisphosphine complex Pd(0)L2 gave almost the same result as the real reaction system. Therefore, we conclude that Pd(0)L2 would be the starting active species in the catalytic cycle, and this conclusion was strongly supported by theoretical calculations (vide infra).
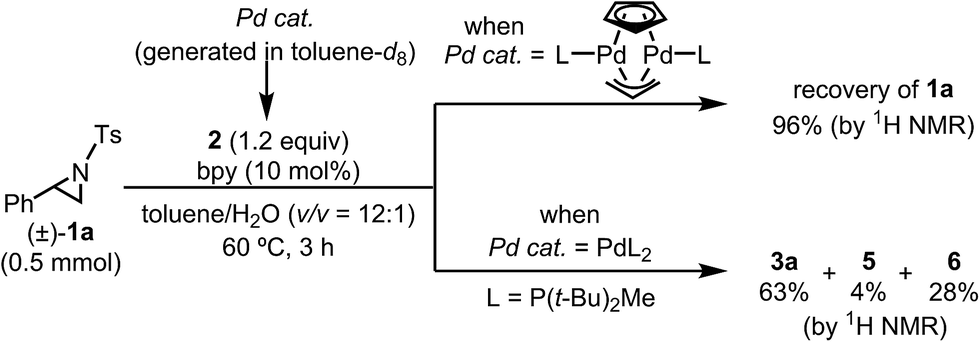 | ||
| Scheme 3 Verification test of the catalytically active species. | ||
Computational part
33–36 functional as implemented in the Gaussian09 program.37 The polarizable continuum model (PCM) was used as the implicit solvent model with a dielectric constant (ε) of 2.3714.38 The SDD basis set and associated effective core potential were applied for Pd,39,40 and 6-31G(d) basis sets were used for the other atoms (BS1).41–44 All stationary points, local minima (LMs) or transition states (TSs), were optimized without any constraint. Vibrational frequency calculations were performed to confirm the nature of the stationary points. Pseudo-IRC calculations were performed to confirm the connectivity between the TSs and LMs. The final potential energies of the optimized structures were calculated as single-point energies on the optimized structures, where a SDD basis set was used for Pd, and cc-pVTZ basis sets were used for the other atoms (BS2).45–47 In the results section, we report both the relative Gibbs free energy (ΔG) at 298.15 K and 1 atm and the relative electronic energy with the zero point energy correction (ΔE).
The aziridine ring opening step of the mechanism takes place via many different TSs, and therefore a proper sampling is very important. An automatic exploration of all important reaction pathways was accomplished using the multi-component artificial force induced reaction (MC-AFIR) method, as implemented in the global reaction route mapping (GRRM) strategy.48–50 An artificial force parameter (γ) of 300 kJ mol−1 was applied, and this is suitable for finding TSs within 300 kJ mol−1. The MC-AFIR calculations were terminated when no new AFIR LM was found for 10 consecutive AFIR paths (NFault = 10). In AFIR calculations, the energy and derivatives were obtained using the ONIOM(B3LYP-D3:PM6-D3) method.51–57 Partitioning of the molecular system is shown in Fig. 2a. A model catalyst was used for MC-AFIR calculations (Fig. 2). SDD basis sets and the associated effective core potentials were used for palladium, and 3-21G basis sets (BS3) were applied for the high-level region of ONIOM calculations.58–60 All AFIR paths were inspected and approximate TSs were identified. Then, the real phosphine ligand was introduced to all approximate TSs, and the TS structures were fully optimized (without artificial force) with B3LYP-D3/BS1 method. A Boltzmann distribution of the transition states was used to calculate the regioselectivity.
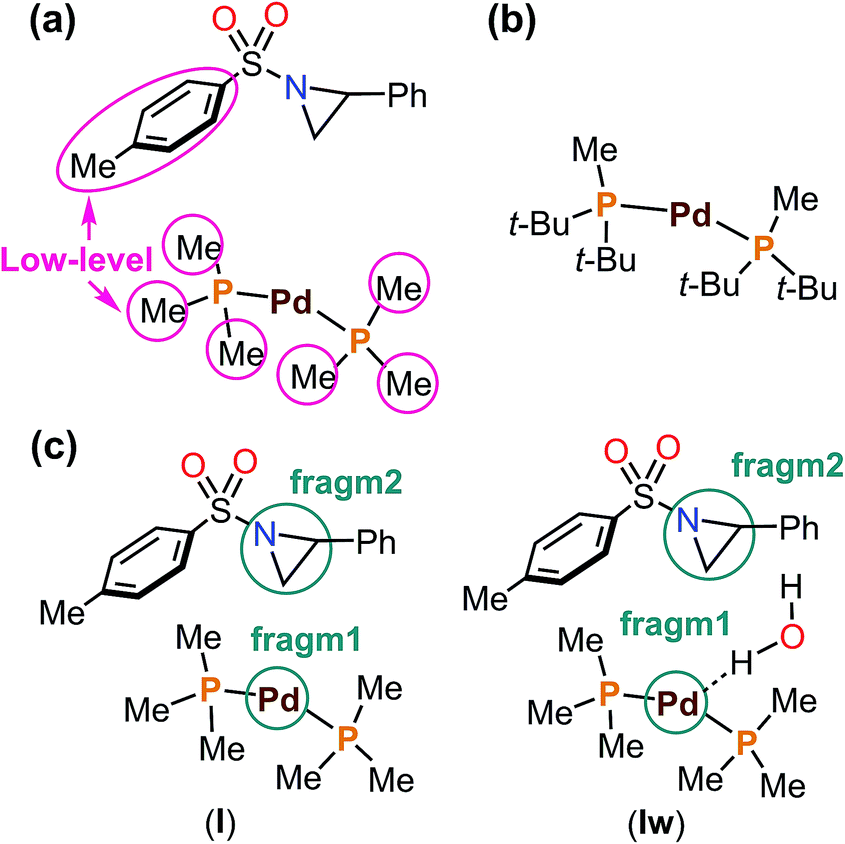 | ||
| Fig. 2 (a) ONIOM partitioning of the model complex into high- and low-levels, (b) the real complex, (c) the artificial force (γ = 300 kJ mol−1) was applied between fragm1 (catalyst) and fragm2 (substrate) without or with an explicit water molecule. | ||
Energy decomposition analysis (EDA)61,62 was performed for the key TSs leading to the desired and the undesired products. B3LYP-D3/BS2 level and PCM were used for the EDA. In this analysis, a TS structure was divided into A (catalyst) and B (substrate) (Fig. 3). INTAB is the interaction energy between A and B at their optimized TS structure. The deformation energy (DEF) is the energy of A and B at the optimized TS, relative to the optimized structures of isolated A and B (denoted as A0 and B0). The energy difference (ΔΔE) between the optimized transition states, (AB)1 and (AB)2, is the sum of the internal energy difference (ΔINTAB) and the deformation energy difference (ΔDEF).
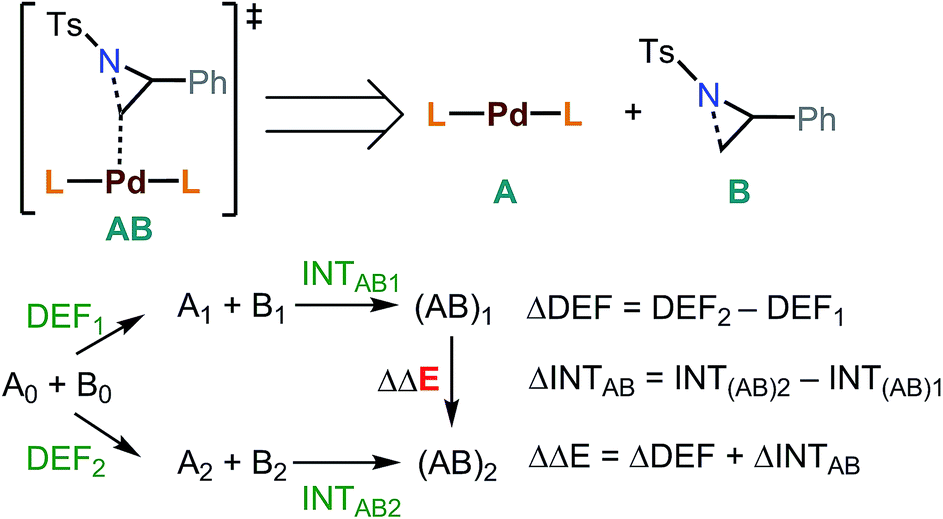 | ||
| Fig. 3 EDA between two optimized transition states leading to the desired and undesired products. | ||
| A | B | ||||
|---|---|---|---|---|---|
| Empty | H2O | bpy | L | Sol | |
| a The symbol “X” indicates that the ligand in parentheses dissociates upon structure optimization. | |||||
| Empty | 0.0, (0.0) | −0.5, (−8.2) | 4.0, (−7.4) | 3.4, (−10.9) | X, (Sol) |
| H2O | — | 4.7, (−14.0) | 2.8, (−17.9) | 7.9, (−15.1) | X, (Sol) |
| bpy | — | — | X, (bpy) | X, (L) | X, (Sol) |
| L | — | — | — | X, (L) | X, (Sol) |
| Sol | — | — | — | — | X, (Sol) |
Starting from the PdL2 complex (I), coordination of one H2O, L, and bpy on I gives rise to PdL2(H2O) (Iw, −0.5 kcal mol−1), PdL3 (3.4 kcal mol−1), and PdL2(bpy) (4.0 kcal mol−1) complexes, respectively. Among the three-coordinate complexes, Iw is the thermodynamically most stable complex, and is only 0.5 kcal mol−1 more stable than I. Therefore, both I and Iw can be formed in solution. In Iw, an H2O molecule coordinates to the metal with one of the two hydrogen atoms. It is important to note that the additive (bpy) can coordinate to the metal, and the resulting complex, PdL2(bpy), is, however, 4.5 kcal mol−1 higher than the most stable complex, Iw. All four-coordinate complexes found in the calculations, PdL2(bpy)(H2O) (2.8 kcal mol−1), PdL2(H2O)2 (4.7 kcal mol−1), and PdL3(H2O) (7.9 kcal mol−1) are relatively higher in energy. Coordination of solvent (Sol) is not possible due to steric repulsion between the bulky groups of the solvent molecules (MTBE) and I. We concluded that the thermodynamically most stable complexes in solution are Iw and I. The general rules that lead to the formation of the thermodynamically stable complexes in solution are: (1) coordination of two ligands on PdL2, which gives rise to four-coordinate complexes, is not favorable; (2) in terms of making three-coordinate complexes, the binding preference of the third ligand follows the order H2O > L > bpy; (3) solvent MTBE would not coordinate to the PdL2 complex due to steric repulsion; (4) coordination of the additive (bpy) is not favorable. According to Table 3, additive coordination on PdL2 is not favorable as the subsequent complex PdL2(bpy) is 4.0 kcal mol−1 higher than PdL2, while the PdL2(bpy)2 complex is not stable. At the same time, it is important to note that aziridine binding on PdL2 is relatively easier (vide infra), and the corresponding adduct is only 2.6 kcal mol−1 higher than PdL2. Therefore, the additive would not coordinate to the PdL2 complex before the aziridine substrate binding. In our present mechanistic study, we do not consider the role of the additive in the mechanism because the reaction works even in the absence of additive (Table 1, entry 4).
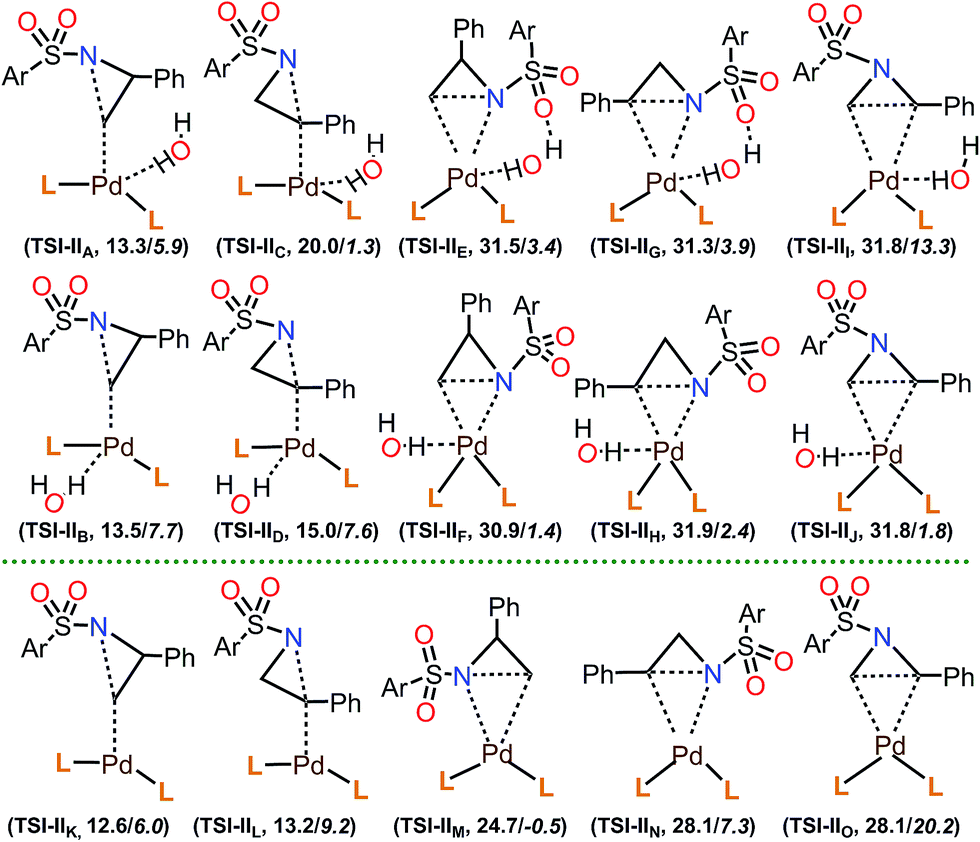 | ||
| Fig. 4 Groups of transition states for the aziridine ring opening, starting from Iw (TSI-IIA through TSI-IIJ) and I (TSI-IIK through TSI-IIO), and energies of the lowest energy TS of each group relative to the PdL2(H2O) (Iw) complex (Ar = p-tolyl; L = P(t-Bu)2Me). ΔG values are in plain text, and ΔE values are in italics. | ||
| TS | Group/regioselectivity | ΔΔG | ΔG (ΔE) | Existence probability (%) |
|---|---|---|---|---|
| a ΔG and ΔE values are indicated in kcal mol−1, relative to Iw. “t” and “b” indicate the terminal (3-position) and benzylic carbons (2-position), respectively. | ||||
| TS1-t-lw | K/terminal | 0.0 | 12.6 (6.0) | 27.6 |
| TS2-t-lw | K/terminal | 0.3 | 12.9 (5.4) | 17.9 |
| TS3-b-lw | L/benzylic | 0.6 | 13.2 (9.2) | 10.0 |
| TS4-t-l | A/terminal | 0.7 | 13.3 (5.9) | 9.6 |
| TS5-t-lw | K/terminal | 0.8 | 13.4 (6.9) | 7.1 |
| TS6-t-l | B/terminal | 0.9 | 13.5 (7.7) | 6.1 |
| TS7-t-l | B/terminal | 1.0 | 13.6 (7.7) | 4.8 |
| TS8-t-l | A/terminal | 1.1 | 13.7 (6.8) | 4.6 |
| TS9-t-lw | K/terminal | 1.3 | 13.9 (7.6) | 2.9 |
| TS10-t-lw | K/terminal | 2.3 | 14.9 (7.6) | 0.6 |
| TS11-b-l | D/benzylic | 2.4 | 15.0 (7.6) | 0.5 |
| TS12-t-lw | K/terminal | 2.6 | 15.2 (−1.2) | 0.4 |
| TS13-t-lw | K/terminal | 2.6 | 15.2 (8.3) | 0.3 |
| TS14-t-lw | K/terminal | 2.7 | 15.3 (7.5) | 0.3 |
| TS15-t-lw | K/terminal | 3.3 | 15.9 (9.0) | 0.1 |
| TS16-t-lw | K/terminal | 3.5 | 16.1 (8.3) | 0.1 |
| TS17-t-lw | K/terminal | 3.6 | 16.2 (7.9) | 0.1 |
| TS18-t-l | B/terminal | 3.7 | 16.3 (9.2) | 0.1 |
| TS19-t-l | A/terminal | 3.7 | 16.3 (−0.8) | 0.1 |
Among the calculated TSs (Table 4), TS1-t-lw (12.6 kcal mol−1) which belongs to the TSI-IIK group is the lowest energy TS of this step. In this TS, the aziridine ring opening takes place at the less hindered carbon in an SN2 fashion, leading to the desired product forming. The same product can be obtained through TS2-t-lw (12.9 kcal mol−1), TS4-t-l (13.3 kcal mol−1), TS5-t-lw (13.4 kcal mol−1), TS6-t-l (13.5 kcal mol−1), TS7-t-l (13.6 kcal mol−1), TS8-t-l (13.7 kcal mol−1), and TS9-t-lw (13.9 kcal mol−1). On the other hand, at TS3-b-lw (13.2 kcal mol−1) of the group TSI-IIL, aziridine ring opening occurs at the hindered carbon (the 2-position), and this is the lowest TS leading to the undesired product. Based on a Boltzmann distribution over the calculated TSs, the regioselectivity is calculated to be 89:11, which is qualitatively in agreement with the experimental results (99:1).
In the calculated TSs for TSI-IIE (31.5 kcal mol−1), TSI-IIF (30.9 kcal mol−1), and TSI-IIM (24.7 kcal mol−1), aziridine ring opening occurs at the less hindered carbon (Fig. 4). However, these TSs are relatively higher in energy due to the fact that the N-p-toluenesulfonyl and 2-phenyl substituents of the substrate are much closer to the bulky groups of Iw or I. Similarly, TSs in the group of TSI-IIG (31.3 kcal mol−1), TSI-IIH (31.9 kcal mol−1), and TSI-IIN (28.1 kcal mol−1) show high energy barriers, where aziridine ring opening occurs at the hindered carbon. TSs in the group of TSI-III (31.8 kcal mol−1), TSI-IIJ (31.8 kcal mol−1), and TSI-IIO (28.1 kcal mol−1) represent the aziridine ring opening through the carbon–carbon bond, which is, however, not possible due to very large reaction barriers. Furthermore, we checked the effect of H2O molecules on aziridine ring opening. Starting from the lowest energy TS for this step (TS1-t-lw, 12.6 kcal mol−1), up to four H2O molecules were introduced, and the corresponding TSs were calculated. The calculated ΔG of TS1-t-lw with one H2O molecule (TS1-t-lw-w, 16.1 kcal mol−1), two H2O molecules (TS1-t-lw-w2, 14.3 kcal mol−1), three H2O molecules (TS1-t-lw-w3, 15.6 kcal mol−1), and four H2O molecules (TS1-t-lw-w4, 16.4 kcal mol−1) suggested that explicit H2O molecules in the system would not stabilize the TS1-t-lw.
TS1-t-lw is a major contributor to the aziridine ring opening at the less hindered carbon, while TS3-b-lw is the lowest TS for aziridine ring opening at the hindered carbon (Fig. 5). The calculated Gibbs free energy difference between TS1-t-lw and TS3-b-lw is 0.6 kcal mol−1, while the potential energy difference is 3.2 kcal mol−1. When we do not consider the zero point energy corrections, the potential energy difference is 3.5 kcal mol−1, and we have used this value for the EDA. According to the EDA (Table 5), the origin of this difference comes from ΔINTAB (3.9 kcal mol−1), indicating better interactions at TS1-t-lw. This is due to the fact that the aziridine substrate can approach closer to the metal in TS1-t-lw (Pd–C = 2.47 Å) (vs. Pd–C = 2.61 Å at TS3-b-lw) (Fig. 5) than in TS3-b-lw due to the lower steric repulsion.
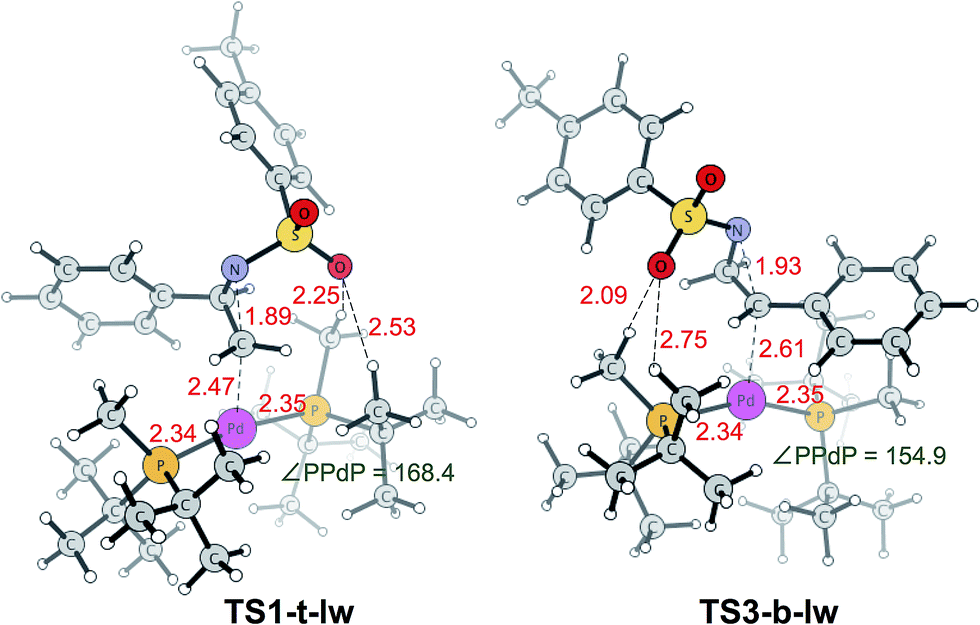 | ||
| Fig. 5 Lowest energy TSs leading to the desired product (TS1-t-lw) and undesired product (TS3-b-lw). Bond lengths (red) are in Å and bond angles (green) are in degrees. | ||
| TS | DEF (DEFA, DEFB) | INTAB | ΔE |
|---|---|---|---|
| TS1-t-lw | 23.2 (1.2, 22.0) | −26.5 | −3.3 |
| TS3-b-lw | 22.8 (2.7, 20.1) | −22.6 | 0.2 |
| ΔDEF | ΔINTAB | ΔΔE | |
| −0.4 (−1.5, 1.1) | 3.9 | 3.5 |
The free energy profile for the early stages of the mechanism is shown in Fig. 6. The reaction starts from the thermodynamically most stable complex, Iw. Therefore, we report energies relative to Iw. Then, aziridine coordination on I leads to a complex II (2.6 kcal mol−1), and the subsequent aziridine ring opening occurs through TS1-t-lw (TSII–III, 12.6 kcal mol−1). Aziridine ring opening is also possible from Iw (not shown in Fig. 6) with an overall barrier of 13.3 kcal mol−1 (TS4-t-lw), which is, however, 0.7 kcal mol−1 higher than TS1-t-lw. Beyond TS1-t-lw, an intermediate, III (8.4 kcal mol−1) is formed.
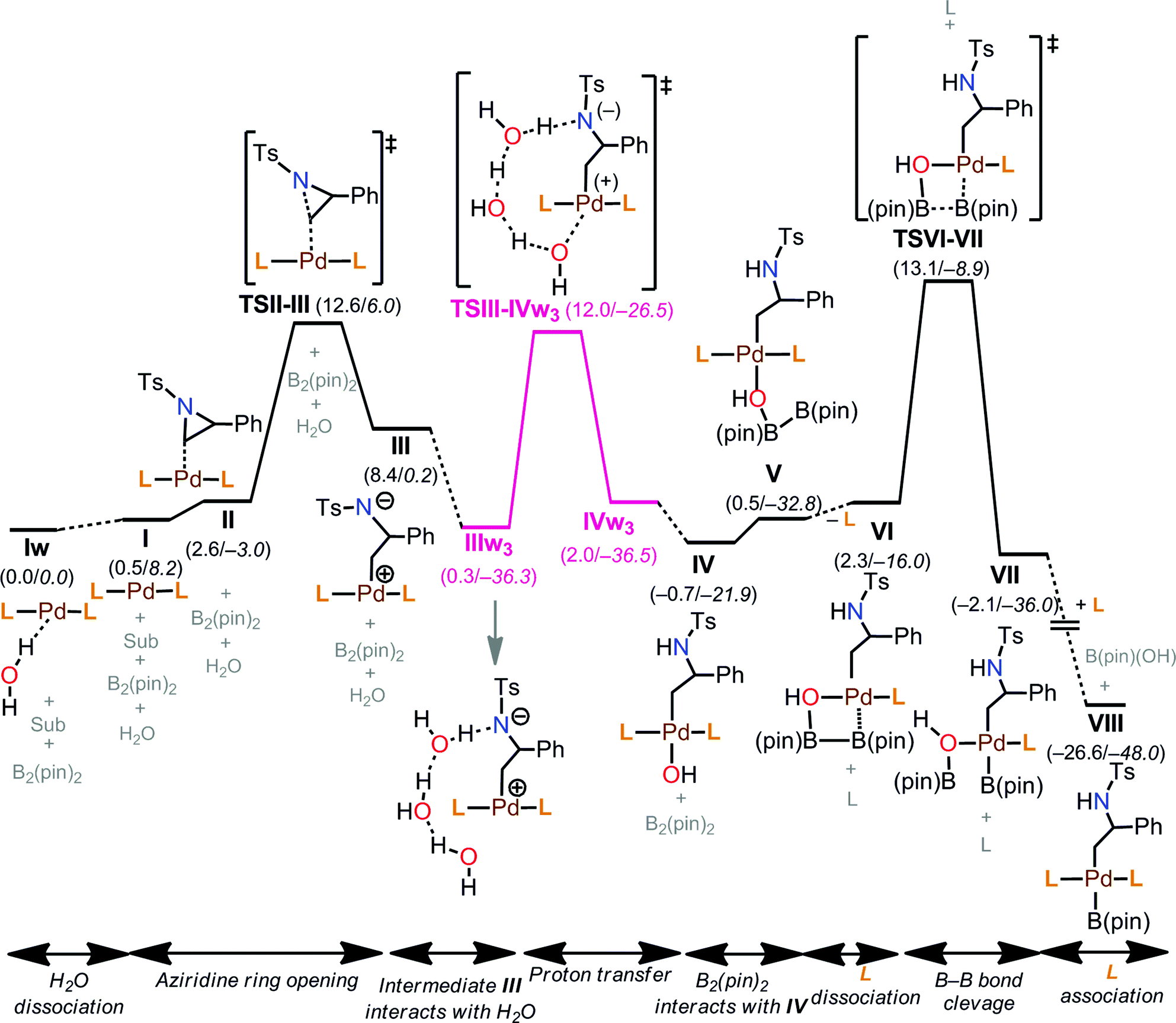 | ||
| Fig. 6 Free energy profile for the reaction mechanism (aziridine ring opening, proton transfer, and B–B bond cleavage processes). Energies are indicated in kcal mol−1. ΔG values are in plain text, and ΔE values are in italics. | ||
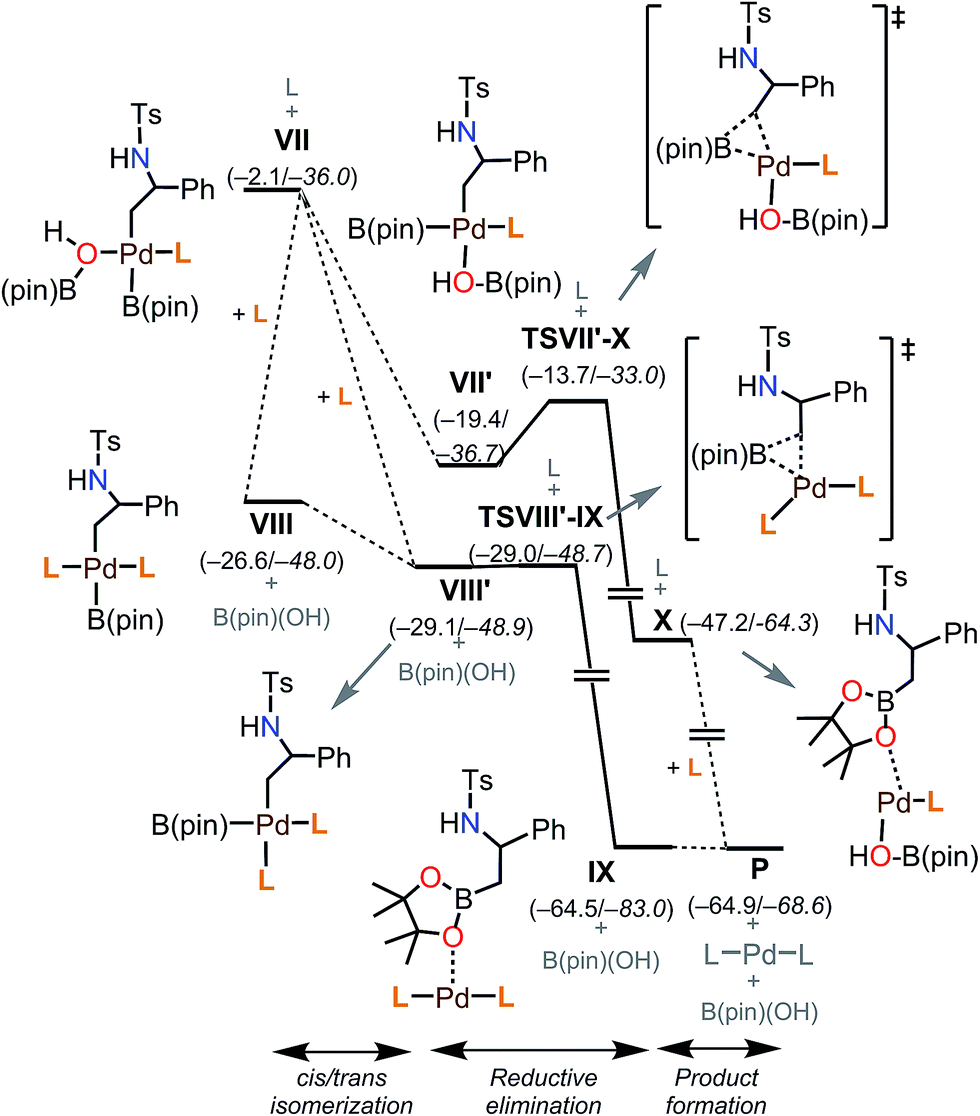 | ||
| Fig. 7 Free energy profile for the later steps of the mechanism. Energies are indicated in kcal mol−1, ΔG values are in plain text, and ΔE values are in italics. | ||
Side product formation
Under the optimized reaction conditions, imine 6 is formed as a side product (Table 1). We have explored the mechanism for the side reaction as shown in Fig. S4 in the ESI.†Catalytic cycle
Putting together the present results from the experimental and theoretical studies, the proposed catalytic cycle for the borylation is shown in Fig. 8 (black cycle). Pd(0)L2 is generated through the sequential reduction of Cp(allyl)Pd. Then, Pd(0)L2 attacks the less hindered carbon of the aziridine in an SN2 fashion (a) to give a stereo-inverted oxidative adduct. A hydrogen bonded chain of H2O molecules plays two roles in the following steps: (i) as a proton source to quench TsN− (b) and (ii) as an internal base to form the [Pd(OH)] species. This intermediate activates the B–B bond of B2(pin)2 (c). The dissociation of a phosphine ligand (d) facilitates the transmetalation (e). The free phosphine ligand participates in the catalytic cycle again to form the (alkyl)PdL2B(pin) complex (f). The trans–cis isomerization (g) followed by reductive elimination (h) leads to the C–B cross-coupled product and the completion of the catalytic cycle. A side reaction may occur after the oxidation addition (brown cycle): the oxidative adduct of aziridine undergoes hydrogen transfer (i) followed by reductive elimination (j) to produce an imine byproduct. Our computational study suggests that the aziridine ring-opening step for substrate 1a has barriers of 12.6 kcal mol−1 (terminal position) and 13.2 kcal mol−1 (benzylic position) (Table 4). For an alkyl aziridine, in particular 1l, the aziridine ring-opening step showed barriers of 15.4 kcal mol−1 (2-position) and 16.3 kcal mol−1 (3-position). In the absence of the aryl group, the aziridine ring opening of 1l is 2.8 kcal mol−1 higher than 1, where the reaction is difficult with 1l. This is qualitatively in agreement with our experimental results, where the reaction does not proceed with 1l.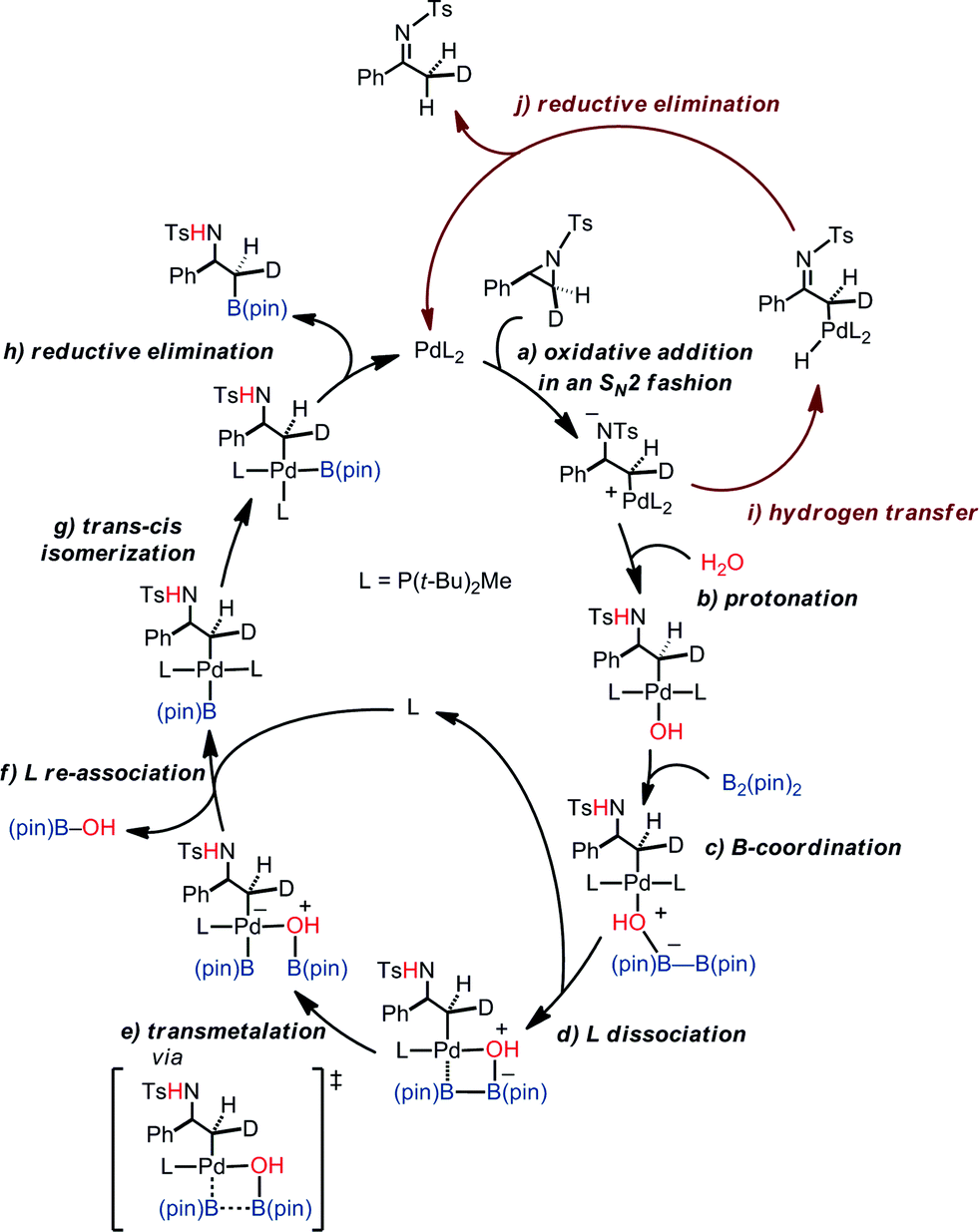 | ||
| Fig. 8 A proposed catalytic cycle of the borylation. | ||
Conclusions
In conclusion, we have developed a Pd-catalyzed regioselective borylative ring opening reaction of 2-arylaziridines to give β-aminoalkylboronates that are otherwise difficult to synthesize using existing methodologies. Importantly, the regioselectivity of the ring opening is controlled by the interactions between the catalyst and the substrate. The SN2 nature of the oxidative addition of aziridine was verified using deuterated aziridine and computational studies. Furthermore, the borylative reaction is applicable under neutral conditions that allow for high functional compatibility. The mechanism of the full catalytic cycle was proposed using DFT and MC-AFIR methods. The aziridine ring opening is initiated by the active species PdL2. TSs were systematically determined for the selectivity-determining aziridine ring opening step, and the calculations reasonably reproduced the experimental regioselectivity. The next step of the mechanism is a proton transfer that is facilitated by a H2O hydrogen bond chain. The resulting Pd-hydroxo species activates the transmetalation step, where inner sphere boron–boron bond cleavage occurs, and leads to the final reductive elimination step. These experimental and theoretical findings open up an avenue to the further development of transition metal-catalyzed ring-opening C–E bond forming cross couplings of aziridines.Acknowledgements
This work was partly supported by a Scientific Research on Innovative Area (JSPS KAKENHI Grant Number JP16H01023 in Precisely Designed Catalysis with Customized Scaffolding, to YT) from MEXT, Japan, and by a Research Grant for the Toray Award in Synthetic Organic Chemistry from the Society of Synthetic Organic Chemistry, Japan (to YT). One of the authors (YT) would like to acknowledge all the warm support from the Frontier Research Base for Global Young Researchers, Osaka University, on the Program of MEXT, Japan. WMCS and KM are grateful to Prof. Satoshi Maeda of Hokkaido University for the developmental version of the GRRM code. WMCS acknowledges JSPS for a Foreign Postdoctoral Fellowship (P14334). This work was in part supported by Grants-in-Aid for Scientific Research (KAKENHI 15H00938 and 15H02158) to KM at Kyoto University. The computer resources at the Institute for Information Management and Communication (IIMC) at Kyoto University and Research Center of Computational Science (RCCS) at the Institute for Molecular Science are also acknowledged.Notes and references
-
Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006 Search PubMed
.
- For reviews on ring opening of aziridines, see:
(a) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599–619 CrossRef
- For a review on the preparation and reactivities of azametallacyclobutanes, see: A. Dauth and J. A. Love, Dalton Trans., 2012, 41, 7782–7791 RSC
- B. L. Lin, C. R. Clough and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 2890–2891 CrossRef CAS PubMed
- J. E. Ney and J. P. Wolfe, J. Am. Chem. Soc., 2006, 128, 15415–15422 CrossRef CAS PubMed
-
(a) H. Alper, F. Urso and D. J. H. Smith, J. Am. Chem. Soc., 1983, 105, 6737–6738 CrossRef CAS
- For a review on the transition metal-catalyzed transformations of small ring compounds including aziridines, see: C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed
- A wide variety of ring expansions of vinylaziridines via π-allylmetal species and ring-opening SN2′ substitutions have been developed. For reviews, see:
(a) E. A. Ilardi and J. T. Njardarson, J. Org. Chem., 2013, 78, 9533–9540 CrossRef CAS PubMed
- Wolfe and coworkers reported a Pd/PR3-catalyzed isomerization of aziridines to ketimines via a tandem process consisting of oxidative addition/β-hydride elimination/tautomerization, see: J. P. Wolfe and J. E. Ney, Org. Lett., 2003, 5, 4607–4610 CrossRef CAS PubMed
-
(a) C.-Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 9541–9544 CrossRef CAS PubMed
-
(a) M. L. Duda and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 18347–18349 CrossRef CAS PubMed
- The Pineschi group reported a Ni(0)-catalyzed borylative ring opening of vinylaziridines that proceeds in a formal SN2′ fashion. Apparently, this reaction proceeds through an oxidative addition of vinylaziridines to Ni(0) to generate a π-allyl Ni(II) species. However, little is described about its mechanism: S. Crotti, F. Bertolini, F. Macchia and M. Pineschi, Org. Lett., 2009, 11, 3762–3765 CrossRef CAS PubMed
-
(a)
Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed
- S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478–10479 CrossRef CAS PubMed
- As a relevant transformation, a metal-free borylative ring opening of vinyl epoxides and aziridines has been reported: X. Sanz, G. M. Lee, C. Pubill-Ulldemolins, A. Bornet, H. Gulyás, S. A. Westcott, C. Bo and E. Fernández, Org. Biomol. Chem., 2013, 11, 7004–7010 Search PubMed
- For selected examples of synthetic methods of β-aminoboronates, see:
(a) D. N. Butler and A. H. Soloway, J. Am. Chem. Soc., 1964, 86, 2961 CrossRef CAS
- For reports on the biological activities of β-aminoethylboronates, see:
(a) A. S. Gorovoy, O. Gozhina, J.-S. Svendsen, G. V. Tetz, A. Domorad, V. V. Tetz and T. Lejon, J. Pept. Sci., 2013, 19, 613–618 CrossRef CAS PubMed
- Recently, Ni-catalyzed benzylic C(sp3)–N borylation of benzylammonium triflates has been reported:
(a) H. Zhang, S. Hagihara and K. Itami, Chem.–Eur. J., 2015, 21, 16796–16800 CrossRef CAS PubMed
-
(a) T. Ishiyama, T. Ahiko and N. Miyaura, Tetrahedron Lett., 1996, 37, 6889–6892 CrossRef CAS
-
(a) H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034–16035 CrossRef CAS PubMed
-
(a) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed
- For detailed information about the effects of the reaction conditions on yields and product distributions, see the ESI†.
-
(a) M. R. Netherton and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 3910–3912 CrossRef CAS
- J. Johnson and T. Rovis, Angew. Chem., Int. Ed., 2008, 47, 840–871 CrossRef CAS PubMed
-
(a) H. F. Bettinger, M. Filthaus, H. Bornemann and I. M. Oppel, Angew. Chem., Int. Ed., 2008, 47, 4744–4747 CrossRef CAS PubMed
- For a review on the cross coupling of alkyltrifluoroborates, see: G. A. Molander, J. Org. Chem., 2015, 80, 7837–7848 CrossRef CAS PubMed
- T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed
-
(a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed
- L. Zhang, Z. Zuo, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 CrossRef CAS PubMed
- L. K. Lukanov, A. P. Venkov and N. M. Mollov, Synthesis, 1987, 204–206 CrossRef CAS
- D. M. Norton, E. A. Mitchell, N. R. Botros, P. G. Jessop and M. C. Baird, J. Org. Chem., 2009, 74, 6674–6680 CrossRef CAS PubMed
- E. A. Mitchell and M. C. Baird, Organometallics, 2007, 26, 5230–5238 CrossRef CAS
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS
- C. T. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS
-
T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry: Methods of Electronic Structure Theory, ed. H. F. Schaefe, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS
- S. Maeda, T. Taketsugu and K. Morokuma, J. Am. Chem. Soc., 2015, 137, 3433–3445 CrossRef CAS PubMed
- S. Maeda and K. Morokuma, J. Chem. Phys., 2010, 132, 241102 CrossRef PubMed
- S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS PubMed
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS PubMed
- T. Vreven and K. Morokuma, J. Comput. Chem., 2000, 21, 1419–1432 CrossRef CAS
- F. Maseras and K. Morokuma, J. Comput. Chem., 1995, 16, 1170–1179 CrossRef CAS
- T. Matsubara, S. Sieber and K. Morokuma, Int. J. Quantum Chem., 1996, 60, 1101–1109 CrossRef
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data of new compounds, copies of NMR and HPLC charts, and calculation results. See DOI: 10.1039/c6sc01120a |
| This journal is © The Royal Society of Chemistry 2016 |