
The functionalization of fluoroelastomers: approaches, properties, and applications
Yu Wang
and
Yongping Bai
*
Department of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, 150001 Harbin, China. E-mail: baifengbai@hit.edu.cn; Tel: +86-451-86413711
First published on 23rd May 2016
Abstract
This review seeks to give an overview of different approaches to prepare functional fluoroelastomers for modern industries. These approaches can be divided into three main families of alternatives. The first extensively describes the preparation of telechelic fluoroelastomers based on chemical degradation. Carboxyl-terminated fluoroelastomers are usually achieved via a typical degradation process, which then can be changed into various functional group-terminated fluoroelastomers. The second deals with functionalization of fluoroelastomers through grafting reactions. Conventional strategies, such as radical polymerization, dehydrofluorination and sulfonation, are used to graft single or faintness functional monomers onto fluoroelastomers. Reversible deactivation radical polymerization, however, provides quantitative copolymers containing unique functional groups. The third alternative route, namely functionalized copolymerization, concerns two main types: functionalization of an initiator and functionalization of monomers. In several cases, both will be utilized to synthesize special functional fluoroelastomers. In addition to the three approaches, the properties and applications of processed fluoroelastomers are illustrated to explore their functions and influences after functionalization.
 Yu Wang | Dr Yu Wang received his bachelor degree in Polymer Material Science and Technology at Harbin Institute of Technology (HIT) in 2010. He continued to pursue a PhD degree under the supervision of Prof Yongping Bai. His main research was focused on the synthesis and applications of functional fluoropolymers. |
 Yongping Bai | Prof Yongping Bai received his PhD degree in Polymer Material Science and Technology at Harbin Institute of Technology (HIT) in 1996. His main research interests include the synthesis and applications of functional fluoropolymers, polyesters and polyacrylate. His research team has successfully developed more than twenty novel products under his direction, including fluorinated coatings, optical BOPET films, flexible polyesters, and special acrylic adhesives. He is also a member of the Editorial Boards of Materials and Design, Materials Letters, Applied Composite Materials and Journal of Applied Polymer Science. |
1. Introduction
Fluorinated polymers always function as specialty materials owing to their unique properties including high thermal, chemical, aging and weather resistance, excellent inertness to solvents, hydrocarbons and acids as well as low dielectric constants, flammability, and moisture absorption.1–5 As one of the most fascinating fluorinated polymers, fluoroelastomers are desirable in many applications that deal with problems of harsh environments and the war industry.3,6–8 These commercial applications include fuel tank hatches, joint sealants, fuel vapor seals, valve seals, hoses, coated cloth, shaft seals and O-rings for hydraulic systems in aircraft and aerospace applications.3,6,9–11 The outstanding performance in these challenging applications inspires extensive research on the synthesis of fluoroelastomers with controlled structures and required functions.12However, commercial fluoroelastomers have some drawbacks. For example, the vulcanization process of fluoroelastomers has to be separated into two steps at high temperature and meanwhile generates a lot of unsaturation sites in the polymer backbone via dehydrofluorination.13 Therefore, the development of simple pathways for the cure of fluoroelastomers has always been a focal point.14–20 Due to the presence of hydrogen bonds in fluoroelastomers, only minor changes in conformational mobility and steric hindrance of C–F hand, and hence, their performances at low temperature are unsatisfactory.21 Moreover, quite a few studies have focused on the blend modification of fluoroelastomers,21–27 so some functional fluoropolymer compatibilizers28–31 should be explored to achieve compatibility32 between fluoroelastomers and other materials. The new functions and new curing chemistry of fluoropolymer materials, as well as the compatibility technologies with conventional materials, can be categorized into the functionalization of fluoroelastomers.
In the past half century, functionalized modification played a positive role in the devolvement of fluoroelastomers. The incorporation of soft segments made it possible for fluoroelastomers to be applied at even lower temperatures, i.e. −50 °C (some products can even keep elasticity at such low temperatures as −80 °C or below).33–37 In some cases, the vulcanization systems and process have even been simplified and conducted at room temperature.16–18,20,38 Additionally, perfluoroelastomers take fluoroelastomers to extreme performance in medium resistance and thermostability.39–41 Fluoroelastomers have also been used to prepare proton exchange or conduct membranes for fuel cells on a trial basis.42–46
Major efforts in early stages focused on copolymerization with functional monomers to adjust the properties of fluoroelastomers. A series of functional fluoropolymers were produced by DuPont, Dow Corning, 3M, Solvay Special Polymers, Asahi Glass Daikin, and ShinEtsu, etc.47–50 To date, copolymerization is still a practical approach to achieve the functionalization of fluoroelastomers in spite of high prices. To lower the cost, exploration for a cost-effective degradation pathway for functional fluoroelastomers has been on the increase since success51,72 with the carboxyl-terminated fluoroelastomers via chemical degradation. Recently, grating reaction also exhibited its advantages in the functionalization of fluoropolymers through controlled polymerizations,52 radical substitution,29 radiation,53 and dehydrofluorination.54
Over the past 20 years, developments in the preparation and applications of commercial fluoroelastomers and fluoroplastics have been summarized in several review articles and books.3,6,33,45,48,49,55–61 Our intention is not to provide another wide spanning review of fluoroelastomers, but rather to describe recent developments of fluoroelastomers from the perspective of functionalized modification. Hence, we attempt to systematically summarize the routes to incorporate functional groups in fluoroelastomers; namely, the functionalization of fluoroelastomers. In order to provide more inspiration for readers, some important references about the functionalization of important fluoropolymers containing chlorotrifluoroethylene (CTFE) and hexafluoropropylene (HFP) are included in the review as well.
2. Functionalization via chemical degradation
2.1 Carboxyl-terminated fluoroelastomers
The chemical degradation of fluoroelastomers is conducted in two main steps: dehydrofluorination reaction and chain scission reaction.62–66 Fluoroelastomers show poor resistance to the alkaline medium,64 which provides a solid foundation for their degraded functionalization. Fluoroelastomers, as one of the halohydrocarbons, tend to eliminate β-H and fluorine atoms in the backbone,67 and then unsaturated bonds are introduced into fluoroelastomers under the alkali's attack. In this process, Hofmann's rule is followed (this is different from other hydrochloric ethers68). After π bonds in unsaturated structures are broken by oxidants, carbonyl groups (with weak oxidants) and carboxyl groups (with strong oxidants) are obtained.69 The mechanism of the oxidative degradation reaction is illustrated in Fig. 1.38 | ||
| Fig. 1 Mechanism of chemical degradation reaction. Reprinted with permission from ref. 38. | ||
It should be noted that not all the vinylidene fluoride fluoroelastomers can be used as raw materials for preparation of carboxyl-terminated fluoroelastomers via chemical degradation. Schmiegel realized that only those fluoroelastomers whose backbones involve sensitive groups (as shown in the following part) to alkali are qualified:70

The molecular weight and carboxyl end group value of carboxyl-terminated fluoroelastomers are significantly dependent on alkaline strength, solvent solubility, and oxidant species. Oxidants can be chosen from H2O2, KMnO4, OsO4, K2Cr2O7–H2SO4, O3, etc. Various alkalis mainly include hydroxide and amines. And solvents such as acetone, butanone, THF, and DMF can dissolve fluoroelastomers.71
Degradable methods can be separated into two alternatives: one-step and two-steps. The first type of method adds both alkalis and oxidants at the same time to achieve carboxyl termination. Mizuide et al.62 and Li et al.38,51 took this pathway so as to degrade commercial fluoroelastomers into low-molecular weight productions. The maximum COOH percentage can reach 3.6% during the process of degradation. But, so far there is no evidence to prove that carboxyl end groups can be found in every fluoropolymer. Carbonyl or aldehyde groups are also distributed at the ends of polymers.
The second alternative is carried out in the following steps: (i) add alkali for dehydrofluorination; (ii) separate fluoride copolymers and add oxidants to accomplish the oxidation reaction. The oxidants used in these types of methods are stronger, compared to the one-step degradation. Cluff et al.72 heated THF mixtures of fluoroelastomers and amines, and then separated and oxidized the products with KMnO4. Similarly, Mizuide73 prepared carboxyl-terminated fluoroelastomers (COOH% = 0.2–3.75%) through the oxidation reaction of KMnO4 or HNO3. Because the Mn element in KMnO4 could cause pollution in the environment, such processes should be well controlled to avoid pollution. So the one-step degradation methods are relatively more moderate, simpler, and more environmentally friendly.
2.2 Other activated group-terminated fluoroelastomers
Different types of telechelic fluoroelastomers can be prepared by virtue of esterification and reduction of a carboxyl (as shown in Fig. 2). Qi's team38,74–79 devoted a good deal of effort to obtain a series of telechelic fluoroelastomers (Fig. 3). Activated pentafluorophenol (PFP),77 silane,78 carborane,77 sulfonyl,74 hydroxyl74,79 and amino74 were introduced via such reactions as the Steglich reaction and Mitsunobu reaction. Different approaches for the same products always result in different conversion percentages. For example, both direct amidation and indirect methods by the Steglich reaction are alternatives to prepare silane-terminated fluoroelastomers, but the carboxyl conversion of direct esterification produces just 71.0%.38 Meanwhile, the carboxyl conversion of indirect esterification using mica ester produces 88%, which is attributed to an easy reaction between PFP and AMEO.38,78 AlLiH4 and NaBH4 are both effective reductants in the conversion reaction to hydroxyl-terminated fluoroelastomers; however, different conversions will be observed. Although AlLiH4 displays such strong reducibility that even the carbonyl of a rearrangement reaction can be reduced to a hydroxyl, no hydration process is required.74 Hence, the application of AlLiH4 in large-scale production is limited. On the other hand, because NaBH4's reducing capacity is rather weak, it can only exhibit its reducing capacity for carboxyl in the presence of a Lewis base.74,79 But the prerequisites for the reaction of borohydride complex systems are rather milder, compared to AlLiH4.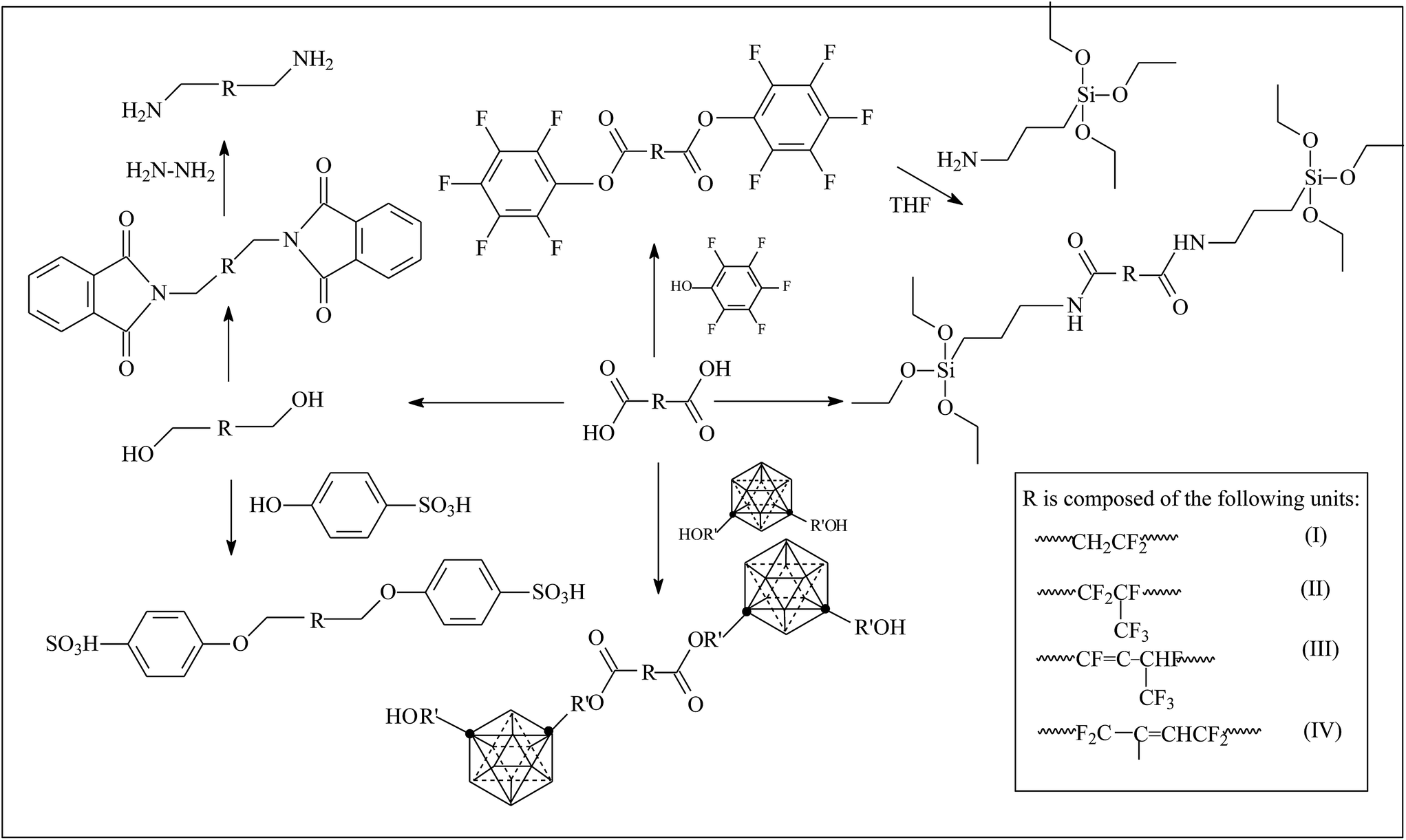 | ||
| Fig. 2 Various routes to preparation of function-terminated fluoroelastomers.38,75–79 | ||
 | ||
| Fig. 3 Grafting mechanism of an aromatic-containing amine onto the poly(VDF-co-HFP) copolymer. Reprinted with permission from ref. 111. | ||
Combinations of chemical degradation and conversion reactions have been proven to be a simple way to obtain a wide range of functional fluoroelastomers. But, some problems still arise. For instance, how to eliminate unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds thoroughly in an oxidation process remains a great challenge in degradation methods. After degradation, functional fluoroelastomers show a low tensile strength. This drawback will reduce their lifetime in their application in shock absorbers.
C double bonds thoroughly in an oxidation process remains a great challenge in degradation methods. After degradation, functional fluoroelastomers show a low tensile strength. This drawback will reduce their lifetime in their application in shock absorbers.
3. Functionalization via grafting reaction
Grafting reaction is a significant pathway to functionalize fluoropolymers by hanging functional groups such as carboxyl,80 hydroxyl,53 sulphonic acid,81 and carbonyl52 onto their backbones. Many methods have been adopted to achieve a large variety of fluoride grafts, including chemical oxidation,54,82–84 free radical polymerization (FRP),29,30 high-energy inducement (HE)53 and several novel chemical modifications.85 Table 1 shows the utilized monomers and corresponding methods for functionalization of different fluoropolymers.| Fluoropolymer | Functional monomer | Grafting technologies | Reference |
|---|---|---|---|
| VDF-HFP |  |
FRP | 29 |
| VDF-HFP |  |
FRP | 30 |
| VDF-CTFE | 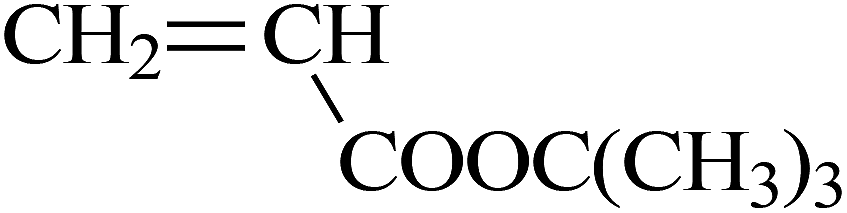 |
ATRP | 52 |
| VDF-CTFE |  |
ATRP | 52 |
| VDF-HFP |  |
O3/O2 treatment | 53 |
| VDF-HFP |  |
Dehydrofluorination | 54 |
| VDF-HFP |  |
Gamma radiation | 80 |
| VDF-CTFE-TBPAC | CH2CF2 |
FRP (macroinitiator) | 86 and 87 |
| VDF-HFP-TFMAA |  |
Mitsunobu reaction | 88 |
| VDF-HFP |  |
Dehydrofluorination | 89 |
| VDF-HFP |  |
Gamma radiation | 90–96 |
| VDF-CTFE |  |
ATRP + sulfonation reaction | 60, 85 and 97 |
| VDF-CTFE |  |
ATRP | 98 |
| VDF-CTFE |  |
ATRP | 99 |
| VDF-HFP |  |
Dehydrofluorination | 100 |
| VDF-HFP |  |
Dehydrofluorination | 101 and 102 |
| VDF-HFP |  |
FRP | 103–106 |
3.1 Grafting via radical polymerization
Grafting silicones from fluoroelastomers via radical grafting is one effective approach for preparing fluorinated silicone materials.107,108 Guo et al. proposed a grafting process which is separated into three sections: the formation of macro radicals, grafting VTEO, and radical transform. After capturing the free radical of VTEO, KFM-g-VTEO is produced and the new KFM macro radical begins to graft new VTEO. Similarly, the same team30,109 also grafted γ-methacryloxypropyltrimethoxysilane to the fluoroelastomers. These two grafted polymers can be used as compatibilizers of a KFM and MVQ blend. But no adequate evidence was presented for the proposed mechanism and grafting ratios were not given in their studies either.In addition to silicones, grafting maleic anhydride via radical copolymerization is another typical modification of fluoroelastomers. Dong et al.105,106 investigated the effects of reaction conditions on the grafting ratio. The highest grafting ratio (1.9%) was obtained when the conditions were set as 0.4 wt% DCP, 10 wt% MAH, and 150 °C. KFM-g-MAH can be utilized to increase compatibilization of fluoroelastomers with acrylic rubber (ACM)103,110 or thermoplastic polyurethane (TPU).105
3.2 Grafting via chemical oxidation
In order to graft functional monomers onto fluoroelastomers via chemical oxidation, expected groups and other groups, such as amino or hydroxyl groups, should be involved in grafted monomers. The grafting process via chemical oxidation contains three steps: (i) eliminate HF in the poly(VDF-co-HFP) copolymer, thus creating double bonds; (ii) subsequently rearrange the dehydrofluorinated poly(VDF-co-HFP) copolymer; and finally (iii) substitute the phenol (or amido) onto double bonds.89Taguet et al.82,111 grafted a series of commercially available amines bearing aromatic rings onto poly(VDF-co-HFP) copolymers. With no methylene spacer, grafting will not be successful. The authors claimed that the higher the molar percentage of HFP, the higher the maximum amount of grafted benzylamine. This may be attributed to the rearrangement of created CFCH bonds (Fig. 3). The synthetic amine salt (H2N–CH2–CH2–S–CH2–CH2–C6H4–SO3Na)54 was also suspended onto unsaturated VDF-HFP chains. In this work, [mercaptan]0/[monomer]0 initial molar ratio should be controlled as with the one above (Fig. 4). Taguet et al.89 also grafted (C10H21)2(CH3)2N+–O–C6H4–SO3–+N(CH3)2(C10H21)2 salt onto fluoropolymers. However, these molar percentages of grafted phenolates were lower than those of grafted H2N–CH2–CH2–S–CH2–CH2–C6H4–SO3Na. Compared to the amine (H2N–CH2–CH2–S–CH2–CH2–C6H4–SO3Na)-grafted copolymers, the phenate sulfonate seems slightly less thermostable. The grafted poly(VDF-co-HFP) copolymers have found their potential applications in conductive membranes.
 | ||
| Fig. 4 Grafting mechanism of 4-hydroxybenzenesulfonic acid onto the poly(VDF-co-HFP) copolymer, previously dehydrofluorinated. Reprinted with permission from ref. 89. | ||
Phenolic hydroxyl groups also have similar effects on amino groups. Moorlag et al.101,102 in their two patents reported a method to form a silane functionalized fluoropolymer by the reaction between organic grafts and fluoropolymer chains. The grafts include a phenol end group, a linking group, and at least one silane end group.
The dehydrofluorination is a necessary step in grafting via chemical oxidation for providing grafting sites. But unsaturated structures have a negative influence on thermodynamic stability and solvent resistance of products.
3.3 Grafting via reversible deactivation radical polymerization
So far, the graft copolymerization from commercial polymers has been mainly accomplished through free radical routes, which unfortunately often produce many homopolymers, resulting in a low grafting density. Reversible deactivation radical polymerization opened a new door for the preparation of functional grafts with well-defined structures.112,113 Recently, grafting copolymerization from fluoropolymers via RDRPs, including ATRP, NMP, and RAFT, has received more and more attention.55,114–116 Considering that alkyl bromides and alkyl chlorides offer the best controlled consequence, pendant secondary halogen atoms help poly(VDF-co-HFP) copolymers and poly(VDF-co-CTFE) copolymers become potential ATRP initiators.The sulfonic group and corresponding sodium salts have been introduced onto fluoropolymers through styrene-controlled grafting polymerization and sulfonation. Zhang and Russell52 performed the synthesis of poly(VDF-co-CTFE)-g-PS and poly(VDF-co-CTFE)-g-PBA graft copolymers with ATRP. Using BPy as ligand, the degree of polystyrene grafting and time exhibited a nice linear relationship (hence first-order polymerization kinetic was observed). The resulting polymer has a polydispersity index of 1.60, which is higher than that of linear polymers synthesized from typical ATRP. Unfortunately, the authors provided no direct evidence to support the viewpoint that grafting started from the cleavage of the C–Cl bond of CTFE.
Additionally, Zhang et al.97 prepared proton exchange membranes by sulfonating fluoropolymer grafted PS (Fig. 5). Resulting membranes showed notably higher proton conductivity than Nafion 112 conditioned upon higher RH (RH > 60%) or low temperature (T < 120 °C).
 | ||
| Fig. 5 Preparation of PVDF-g-SPS graft copolymers. Reprinted with permission from ref. 97. | ||
Tsang et al.81 compared poly(VDF-co-CTFE)-g-SPS and poly(VDF-co-CTFE)-b-SPS, which were synthesized by ATRP. Authors found that graft copolymers yielded membranes tolerating much higher ionic contents. The authors also demonstrated that nanoarchitectured morphology plays an essential role in determining the properties of proton conducting membranes.
Differing from the above-mentioned approaches, Kim et al.98 grafted PSSA from fluoropolymers via ATRP (Fig. 6) and then crosslinked the poly(VDF-co-CTFE)-g-PSSA under UV irradiation. Interestingly, after introducing Na+, the resulting grafts can be utilized as proton conducting electrolyte membranes, instead of proton exchange membranes. The same team prepared another similar membrane based on the poly(VDF-co-CTFE)-g-PHEA graft copolymer. The membrane's maximum conductivity achieved 0.015 S cm−1, which was lower than the former one. This may be attributed to an incomplete esterification reaction.
 | ||
| Fig. 6 Graft copolymerization of poly(VDF-co-CTFE)-g-PSSA. Reprinted with permission from ref. 98. | ||
Nowadays, atom transfer radical polymerization (ATRP) has been applied to introduce functional groups into fluoropolymers, especially poly(VDF-co-CTFE) copolymers. Although the research on ATRP is not completely robust, the graft ratio and solid content are satisfactory. Therefore, there is tremendous value in the functionality of special fluoropolymers containing fluoroelastomers.
3.4 Grafting via high energy methods
Fluoropolymers are relatively susceptible to high energy methods, despite their high chemical stability.117,118 Functionalized modification of fluoroelastomers by these high energy methods has enhanced some properties.119Plasma treatment is a commonly used method to change the properties of fluoropolymers by introducing functional groups onto their surfaces. Urushihara and Nishino120 modified thermoplastic fluoroelastomers (TPFEs) with O2 plasma treatment. The TPFE is hydrophobic (θ = 96°), and it turned hydrophilic (θ = 36°) after plasma treatment. More importantly, a high hydrophilic surface can be maintained during the stretching/recovering process. After O3/O2 treatment, Rahimabady53 performed thermally-initiated graft copolymerization of DMA (dopamine methacrylamide) from the ozone-pretreated poly(VDF-co-HFP) copolymers (graft ratio = 5.9%) in order to be applied in high energy density storage (E = 33 J cm−3). This work demonstrated that grafting an appropriate amount of polymer with polar groups is an effective strategy for improving polymer dielectrics. On the contrary, Gao et al.121 grafted a great number of hydrophobic groups (most of which were CF3, CF2, and CF) onto the surface of a fluoroelastomer by direct fluorination with fluorine gas. According to the contact angle measurement, no apparent improvement was found in the hydrophobic property of fluoroelastomers.
Similar to plasma treatment, irradiation of fluoroelastomers results in a grafting reaction or the formation of a three-dimensional network through the union of the macro radicals generated. Dutta et al.122 investigated the influence of some polyfunctional monomers including tripropylene glycol diacrylate (TPGDA), trihydroxymethyl propane triacrylate (TMPTA), trimethylolpropane trimethacrylate (TMPTMA), tetramethylene maleic anhydride (TMMA) and triallyl cyanurate (TAC) on fluorocarbon elastomers with the presence of an electron beam. The results showed that 50 kGy is a key radiation dose (beyond this value, oxidation, dehydrofluorination, grafting, crosslinking, and scission change marginally). Machado's group123 discovered that electron beam (EB) irradiation could improve the tensile strength, hardness, and stiffness of fluoroelastomers. A micrograph of the sample, which is irradiated with the highest applied dose (250 kGy), presented a homogeneous surface (as shown in Fig. 7). Recently, swift heavy ion irradiation was used to functionalize poly(VDF-co-HDF) nanohybrid membranes for fuel cell applications.46 After radiation treatment, the bulk dc conductivity of the functionalized membrane increased 13 orders of magnitude, and activation energies also increased to 32 kJ mol−1.
 | ||
| Fig. 7 SEM micrographs of fractured surfaces from: (A) non-irradiated sample and (B) sample after EB irradiation processing up to 250 kGy. Reprinted with permission from ref. 123. | ||
4. Functionalization via copolymerization
4.1 The initiator functionalization
Ameduri's group made a big contribution to the synthesis of fluoropolymers (especially fluoroelastomers) by iodine transfer radical polymerization in the presence of C6F13I. Boyer et al.132 in this team performed the synthesis of poly(VDF-co-PMVE) copolymers using C6F13I as CTA, which controlled the molecular weight of products, ranging from 2000 to 4450 g mol−1. These copolymers showed an amorphous behavior and potential use for elastomer applications.
In addition, the authors12 adopted a similar strategy to successfully introduce a SF5 group into (VDF-ter-HFP-ter-SF5M) terpolymers. When using CF2CFSF5 as a third monomer, it did not terpolymerize under ITP conditions, leading to poor yields. Many other iodine transfer agents, such as C2F5(CF2CF2)I and (F3C)2CFI, have been utilized in some literature135 to prepare various fluoropolymers via telomerization of VDF, HFP and other fluorinated monomers (Fig. 8).
 | ||
| Fig. 8 Direct or sequential co-telomerization of vinylidene fluoride (VDF) or 3,3,3-trifluoropropene (TFP) in the presence of 2-iodoperfluoroisopropane. Reprinted with permission from ref. 135. | ||
Compared to RFI, fluorinated telechelic diiodides (I–RF–I) have been used more frequently to design well-architectured thermoplastic fluoroelastomers. The synthesis of block fluoropolymers from the I–RF–I was pioneered by Tatemoto's group133,134 and Ameduri's group.55,132,135,136 In the past five decades, iodine transfer polymerization of fluoroalkenes has contributed to extensive research on the synthesis of fluorinated hard-b-soft-b-hard triblock copolymers. In this method, fluoroalkenes were usually copolymerized with ethylene to form hard block chains, such as poly(E-alt/co-TFE),137,138 poly(E-co-CTFE),139 poly(E-co-HFP-co-TFE)140 and poly(E-co-HFP-co-PMVE).141
Recently, in the synthesis of telechelic diiodopoly(VDF-co-PMVE) polymers35,132,142 using IC6F12I and IC4F8I, experimental Mn increases linearly with monomer conversion, revealing the controlled or “pseudo-living” character of ITP, VDF, and PMVE. Boyer et al.35 synthesized fluorinated thermoplastic elastomers via a two-step procedure. In the second step, TFE was introduced to establish the hard chain of fluoroelastomers. IC6F12I was utilized again by Sawada et al.143 to act as a chain transfer agent in the ITP of α-trifluoromethacrylic acid with VDF and HFP.
Although iodine-terminated fluoroelastomers have become important telechelic fluoropolymers, terminal iodine is not useful for crosslinking of polymers; it is expected to be transformed into other active groups. DuPont researchers transformed terminated iodines to telechelic bis(cyanate)s, bis(azido)s,144 bis(ester)s, or bis(trialkoxysilane)s.145 And Kostov et al.146,147 provided some strategies to synthesize photo-cross-linkable telechelic diacrylate poly(VDF-co-PMVE) co-oligomers by changing iodines into “acrylate groups” (Fig. 9). When using acryloyl chlorides as acrylating agents, the transformation was most successful. Some reviews report various approaches through which transfer agent RFI is transformed into useful derivatives, which are useful for producing innovative telechelic fluoropolymers (Fig. 10). However, it is still worthwhile to make many efforts to simplify post-processing and improve the yield of final products.
 | ||
| Fig. 9 Overall strategies to synthesize photo-cross-linkable telechelic diacrylate poly(VDF-co-PMVE) co-oligomers. Reprinted with permission from ref. 146. | ||
 | ||
| Fig. 10 Various fluorinated products from perfluoroalkyl ethyl iodide. Reprinted with permission from ref. 126. | ||
| Peroxide initiator | Initiated monomer | Reference |
|---|---|---|
 |
VDF/CTFE | 148 |
 |
VDF/CTFE | 148 |
| H2O2 | VDF/HFP | 149–151 |
| VDF/CTFE | ||
 |
VDF/CTFE | 148 and 152 |
 |
VDF/CTFE | 148, 153 and 154 |
| VDF/PSFVE | ||
 |
VDF/HFP | 155–157 |
 |
VDF/CTFE | 158 |
 |
VDF/HFP/PS | 159–163 |
| VDF/PS | ||
| VDF/MAA | ||
| VDF/BMA |
 | ||
| Fig. 11 Synthesis of BPO-based functional initiators. Reprinted with permission from ref. 149. | ||
 | ||
| Fig. 12 Radical co-polymerization of VDF and HFP by hydrogen peroxide. Reprinted with permission from ref. 151. | ||
Benzoyl peroxide (BPO) has demonstrated its efficiency in radical polymerization; therefore, synthesis of BPO-based functional initiators is a convenient strategy for the chain-end functionality of fluoroelastomers. Wang's group developed routes to chain-end functionalization of fluoropolymers with BPO-functional initiators. The team prepared a series of BPO-functional initiators by utilizing functional benzoic acids and benzoyl chlorides. Zhao et al.149 took advantage of BPO-functional initiators to initiate the copolymerization of VDF and CTFE. The molecular weight of resulting products was 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 or 56000 g mol−1, while the PDI ranged from 1.5 to 1.6. If BPO is functionalized with halogen, this traditional co-polymerization can be combined with ATRP for preparation of novel block fluoropolymers. Most importantly, these functionalized approaches provide new possibilities for the self-assembly of inorganic nanoparticles in a fluoropolymer matrix to form hybrid materials.148
000 or 56000 g mol−1, while the PDI ranged from 1.5 to 1.6. If BPO is functionalized with halogen, this traditional co-polymerization can be combined with ATRP for preparation of novel block fluoropolymers. Most importantly, these functionalized approaches provide new possibilities for the self-assembly of inorganic nanoparticles in a fluoropolymer matrix to form hybrid materials.148
Controlled by functional borane. The success of reversible deactivation radical polymerization by functional borane initiators propelled Chung et al.164,165 to explore the functionality of fluoropolymers. Using this controlled manner, Chung's team166 synthesized poly(VDF-co-CTFE) copolymer and then introduced silicon-containing monomers to form terpolymers which resulted in a fair yield. Five years later, Zhang et al.167 in Chung's team prepared silane-terminated PVDF by silane/borane-terminated initiators. The resulting products can function as an effective polymeric surfactant, which exhibited high interfacial activity in exfoliated fluoropolymer/clay nanocomposites. Nowadays, borane/oxygen radical initiators have been proven to be effective for these fluoromonomers, such as VDF, CTFE and TrFE.
Controlled by functional xanthate. Co-polymerization of VDF and HFP by this method was carried out for the first time by Severac168 in 2003. Two years later, an original fluorinated xanthate was also synthesized by Sauguet.169 Poly(VDF-co-HFP) copolymers were terminated by CH3OCOCH (CH3)SC(S)OEt. When the proportion of VDF and HFP changed at the second step, fluoropolymers can exhibit different chain properties. Some reviews55 also have summarized controlled polymerizations by xanthates, so we didn't present the application of xanthates in the functionality of fluoroelastomers.
4.2 Co-monomer functionalization
In general, except for Viton, the fluorine content of fluoroelastomers ranges from 66.6% to 75.0%, which limits their surface properties. Therefore, it is necessary to incorporate more special groups to optimize fluoroelastomers' properties.170,171 The pentafluorosulfanyl (SF5) group shows outstanding properties, such as a good lubricant, insulating properties, and oil-resistance.172,173 Ameduri's team12 reported the terpolymerization of both SF5-monomers (i.e., F2CCFSF5 and H2CCHSF5) with VDF and HFP in the presence of C6F13I (see the earlier discussion on functional initiators).
To satisfy the demand of fuel cell technology, novel proton-conducting fluoropolymers were prepared by incorporating aryl sulfonic acids via direct or indirect copolymerization. Through the terpolymerization with VDF and HFP, trifluoromethacrylic acids were introduced into a chemical structure, and then were changed into aryl sulfonic acids via the Mitsunobu reaction.88,143 Sulfonated triblock fluorocopolymers can also be obtained by virtue of the ATRP of fluorinated monomers (mainly including VDF, HFP, and PMVE) and sulfonated monomers.174,175 But researchers prefer to obtain sulfo groups through the sulfonation of styrenes.92,97,159
Interestingly, a few grafted fluoroelastomers can be achieved from radical copolymerization of fluorinated monomers. Ameduri's team prepared fluorinated graft copolymers bearing perfluoropolyether via the terpolymerization of VDF, HFP, and a ω-allyl amide type perfluoropolyether.176,177 Unfortunately, the incorporated amount of allylic perfluoropolyether (PFPE) is poor, so it is necessary to increase the reactivity of this allyl derivative. Similar methods also have been used with pendant ester groups or nitrile groups as crosslinking points of fluoroelastomers. In this way, it is feasible to cure fluoroelastomers at low temperature.19,178
In the last century, fluorosilicone elastomers were prepared by many researchers and companies48,179,180 by using fluorinated groups to replace grafted methyl of polysiloxane macromers. These “fluoroinorganic” elastomers retained good tensile properties at high temperature, and showed exceptional low-temperature flexibility. Shin-Estu pendanted fluorinated chains into main or side chains and synthesized a series of hybrid fluorinated homopolymers.37,151,181 Some novel fluoropolymers were obtained by the radical polymerization of fluorocarbons and silicones in scCO2.20,182–186
In contrast to other commercial polymers, polyurethane exhibited lower Tgs, ranging from −50 to −100 °C. So, the family of polyurethane fluoroelastomers makes up for the shortcoming of KFM's low-temperature resistance. Some articles and patents reported a class of PFPE elastomers.33,187,188 After functionalizing polyurethane fluoroelastomers189,190 with reactive groups, vulcanization is much easier. For instance, Turri's team36 introduced allyl type functions –CH2–CHCH2 into polyurethane fluoroelastomers, so vulcanization can be more easily conducted by the destruction of double bonds in the presence of peroxides (reaction process is shown in Fig. 13).
 | ||
| Fig. 13 Reaction process for the preparation of polyurethane fluoroelastomers. Reprinted with permission from ref. 36. | ||
In addition to fluorosilicone and polyurethane fluoroelastomers, F-phosphazenes also exhibit a low glass transition temperature (Tg = −70 °C). These fluorinated phosphazene elastomers are mainly obtained by sequential polycondensation of phosphoranimines or by hydrosilylation reaction.191,192
5. The properties influence of functionality
5.1 Vulcanization
A hot research topic has always been to vulcanize fluoroelastomers by a simpler process or at a lower temperature.193–197 In order to make fluoroelastomers better curable by peroxides, bromine-containing monomers have been introduced to provide curing sites.198 The progress in curing chemistry of fluoroelastomers has been triggered by the synthesis of various telechelic polymers.38,75,76,80 In this way, terminated carboxyl38 and hydroxyls74 of fluoroelastomers can be vulcanized by isocyanate at room temperature. Epoxy is another effective curing agent for carboxyl-terminated fluoroelastomers, but it requires a higher temperature than 100 °C.38 Under UV radiation, the network can be established from the double bonds of telechelic poly(VDF-co-PMVE) polymers.146 Wang's group148 induced the cross-linking of the amine-terminated fluoropolymers by trimesic acid trichlorides at a relatively low temperature. More conveniently, silane-terminated fluoroelastomers can be cured at room temperature in the presence of DBTDL/TEOS.20,75,80 RGO, MWNT, and POSS have been incorporated for modifying vulcanization behavior or other properties of fluoroelastomers.Additionally, functionality via a grafting reaction and functional monomers copolymerization graft ester groups onto fluoroelastomers. The grafts can provide active posts for polyamide to complete the curing process at room temperature.16–18 Nowadays, click reactions using azide–nitrile cycloaddition also have vastly increased in the vulcanization field.19,174
5.2 Thermal properties
Fluoroelastomers exhibit outstanding resistance to high temperature but poor resistance to low temperature due to their strong fluorinated moiety. Van Cleeff pointed out that the higher the hydrogen contents, the lower the Tgs of Viton elastomers (all their Tgs are higher than −29 °C).199,200 Thermal properties of functional fluoropolymers are dependent on molecular weights and introduced functional groups. The Tg of classical carboxyl-terminated fluoroelastomers, degraded by Qi's team, is about −25 °C,38 which is lower than that of original VDF-HFP fluoroelastomers. Meanwhile, the initial degradation temperature of these liquid fluoroelastomers appears to be at 250 °C.38,80 The thermostability of grafted fluoroelastomers reduces because of fewer fluoride groups and lower molecular weights. The melting points of the CEFRAL® poly(VDF-co-CTFE)-g-PVDF suggested by Central Glass Company Ltd. were 155–160 °C.86,87 The content ratio of soft and hard chains also plays a decisive role in determining thermal properties. For instance, fluoropolymers initiated by H2O2 exhibited lower Tgs (−41 to −77 °C).151 The Tg values of poly(VDF-ter-HFP-ter-CF2CF(SF5)) terpolymers, which were prepared by Kostov et al., ranged from −52 to −42 °C.12 This fluoropolymer showed potential applications as an elastomer because of its amorphous behavior. Furthermore, compared to other functional fluoroelastomers, the super flexibility of PFPE33,36 and silane chains20,48 enables Tgs of polyurethane fluoroelastomers and fluorosilicone to decrease to almost −100 °C.5.3 Surface properties
In general, the surface properties of telechelic fluoropolymers mainly depend on the molecular weight, not the groups at the end of polymers. The contact angles of water on saline-terminated fluorinated elastomer membranes is 91–114°,75,80 which indicates that their hydrophobicity is similar to the original fluoroelastomers' hydrophobicity. On the other hand, the surface energy of grafted fluoroelastomers is mainly dependent on the introduced groups. By direct fluorination, the introduced fluorinated groups enabled fluoroelastomers to increase from 97° to 107°.121 On the contrary, O2 plasma treatments changed hydrophobicity of fluoroelastomers into hydrophilicity (the water contact angle decreased from 96° to 36°).120 When functionalizing via copolymerization with other functional monomers, the surface energy of fluoroelastomers decreases with the presence of hydrophobic monomers. On fluorosilicone coatings20 prepared by Baradie and Shoichet, the copolymerized PDMSMA led to an increase of the water contact angle from 105° to 125°. Enriched SF5 groups exert similar hydrophobic effects on the surface of fluoroelastomers.5.4 Mechanical properties
Table 3 shows the mechanical properties of fluoroelastomers after functionality. In general, the tensile strength and elongation of KFM are 9.0–18.6 MPa and 100–500%, respectively. After degradation, most functional fluoropolymers keep a good elongation but exhibit a lower tensile strength. Especially, the tensile strength of carboxyl-terminated fluoropolymers is just 1.7 MPa. This is attributed to the decrease of precursors' molecular weight (functional grafting and copolymerization also have these drawbacks if the number of suspended activated points is too few or the molecular weight is too low). DuPont has designed a low molecular weight fluoroelastomer using VDF, HFP, and fluorinated acrylate. When the molecular weight of this fluoroelastomer is 4000, its tensile strength is just 1.3 MPa. Grafted fluoroelastomers can be used as compatibilizers in a KFM and VMQ blend. Composited elastomers can only show their best mechanical properties when adding an appropriate content (about 10 phr). In a family of polyurethane fluoroelastomers, diol extender plays a positive role in elongation. In Turri's work, the biggest elongation of polyurethane fluoroelastomers is 490%, which nearly reaches the maximum of KFM's elongation. Although fluoroelastomers show lower surface energy after treatment with fluorine gas, they keep their original mechanical properties.| Functional fluoropolymer | Tensile strength (MPa) | Elongation at break (%) | Shore A hardness | Reference |
|---|---|---|---|---|
| a a and b: mechanical properties correspond to the FKM and MVQ blend. | ||||
 |
1.3–2.5 | 210–300 | 18 | |
| TFE-ter-VAc-ter-PDMSMA | 86 | 350 | 20 | |
 |
8.1–9.0 | 394–427 | 62–64 | 29 |
 |
5.2–8.8 | 440–520 | 30 | |
 |
81–492 | 42–62 | 36 | |
 |
1.77 | 440 | 52 | 38 |
 |
0.22–1.35 | 170–270 | 43–50 | 74 |
 |
1.44–4.45 | 50–300 | — | 75 |
 |
7.32–9.60 | 50–75 | — | 77 |
 |
28/30 | 520/563 | 86 and 87 | |
 |
7.6–81.6 | 8.7–26.6 | — | 98 |
 |
26.2 | 23.4 | 99 | |
| Direct fluorination with fluorine gas | 13 ± 2 | 209 ± 20 | 68 ± 1 | 121 |
6. Applications
Commercial fluoroelastomers are usually utilized as seals,201 rings, coatings,202 gaskets, hoses, expansion joints, etc. in aircraft, aerospace, and car industries. Functional fluoroelastomers overcome some shortcomings of commercial fluoroelastomers, so they are used in a wide range of applications.After degradation, functional fluoroelastomers turn into liquids from solids so that they can adhere readily to many substrates. They are used as protective coatings on elastomers, metals, and other surfaces that need to be protected from harsh service conditions. Interestingly, the decrease of molecular weight, increase in the pressure sensitivity of fluoropolymers, and the resulting fluoropolymers may become novel PSAs. Degradative fluoroelastomers can find applications in many other fields, such as aerospace, automotive, dry cleaning, and high-speed industrial printing industries, plus environmental protection.
Additionally, introduction of silicon increases the flexibility of fluoroelastomers so that they can be used as seals at low temperatures. But, they are less stable at high temperatures than commercial fluoroelastomers. Polyurethane fluoroelastomers also can achieve materials with low temperature or high working pressure, as in the case of a new generation of diesel engines.
After grafting silicone or acrylate, functional fluoroelastomers can act as a compatibilizer of KFM with other elastomers such as MVQ, ACM, or TPU. For instance, by using grafted fluoroelastomers to enhance compatibility of KFM/MVQ, blend rubbers can show similar performances to fluorinated polysiloxanes but at a lower cost.
Some fluoropolymers in the form of VDF-HFP and VDF-CTFE can be developed as proton conducting membranes by incorporating ionogenic functions. And the successful introduction of sulfo groups enables them to function as proton exchange membranes.
7. Conclusions
As the most charming materials, fluoroelastomers have shown their irreplaceable status in many applications. But fluoroelastomers have some drawbacks, such as high price, complex vulcanization, poor resistance at low temperatures and poor compatibility with other components. In order to overcome these drawbacks, more and more attention has been paid to functionalized approaches for obtaining novel structures and better properties.All the functionalized approaches can fall into three main categories: (i) functionalized degradation. Various functional groups are introduced at the end of polymer when the molecular weight decreases. (ii) Functionalized grafting. FRP, oxidation, dehydrofluorination, sulfonation, ATRP, etc. are involved in this method to graft functional groups or functional monomers into the backbone of fluoropolymers. (iii) Functionalized copolymerization. Functional monomers and initiators are used for the preparation of novel functional fluoroelastomers. In fact, these three categories are interrelated. Sometimes, the same functional fluoropolymers can be prepared via different functionalized pathways.
After the introduction of curable groups, quite a few fluoroelastomers can be vulcanized at a relatively low temperature, even at room temperature. A decrease in molecular weight enhances the compatibility of fluoroelastomers with other polymers, and then some nanocomposites can be prepared. Furthermore, embedding soft chains into fluoropolymers improves their resistance at low temperatures. The functionalization of fluoroelastomers enables them to be more widely applied.
However, there are some shortcomings in these functionalized approaches. When using functionalized degradation, too many double bonds and unexpected groups will be introduced into the backbone of a fluoropolymer, which could reduce fluoropolymers' heat resistance. Grafting via dehydrofluorination also faces the same challenge. Grafting via FRP will produce many homopolymers, and grafting via ATRP is not suitable for most fluoropolymers in the form of VDF-HFP. Functionalized copolymerization is expensive, because functional monomers or functional initiators have to be prepared.
Further works are expected to improve functional degrees, decrease unexpected structures, and reduce costs of functional fluoroelastomers. These goals are so attractive that many researchers will devote themselves to this interesting and challenging area.
Abbreviations
| ACM | Acrylic rubber |
| AlLiH4 | Lithium aluminium hydride |
| AMEO | Aminopropyltriethoxysilane |
| ATRP | Atom transfer radical polymerization |
| BMA | Butyl methacrylate |
| BPO | Benzoyl peroxide |
| CTFE | Chlorotrifluor ethylene |
| DBTDL | Dibutyltin dilaurate |
| DCP | Dicumyl peroxide |
| E | Ethylene |
| EB | Electron beam |
| FRP | Free radical polymerization |
| HDI | Hexamethylene diisocyanate |
| HFP | Hexafluoropropylene |
| ITP | Iodine transfer polymerization |
| MAA | Methacrylic acid |
| MAH | Maleic anhydride |
| MVQ | Methyl vinyl silicone rubber |
| MWNT | Multiwalled carbon nanotube |
| NaBH4 | Sodium borohydride |
| NMP | Nitroxide mediated polymerization |
| PFP | Pentafluorophenol |
| PFPE | Perfluoropolyether |
| PFSVE | Perfluoro(4-methyl-3,6-dioxane-7-ene) sulfonyl fluoride |
| PMVE | Perfluoromethyl vinyl ether |
| POSS | Polyhedral oligomeric silsesquioxane |
| PS | Polystyrene |
| PSSA | Poly(styrene sulfonic acid) |
| RAFT | Reversible addition–fragmentation chain transfer |
| RDRP | Reversible deactivation radical polymerization |
| RGO | Reduced graphene oxide |
| RF | Perfluorinated group |
| RFI | Perfluoroalkyl iodide |
| scCO2 | Supercritical carbon dioxide |
| TAC | Triallyl cyanurate |
| TBPAC | tert-Butyl peroxyallyl carbonate |
| TEOS | Tetraethyl orthosilicate (or ethyl silicate or tetraethoxysilane) |
| TFE | Tetrafluoroethylene |
| TFMAA | 2-(Trifluoromethyl) acrylic acid |
| THF | Tetrahydrofuran |
| TMMA | Tetramethylene maleic anhydride |
| TMPTA | Trihydroxymethyl propane triacrylate |
| TMPTMA | Trimethylolpropane trimethacrylate |
| TPFE | Thermoplastic fluoroelastomers |
| TPGDA | Tripropylene glycol diacrylate |
| TPU | Thermoplastic polyurethane |
| TrFE | Trifluoroethylene |
| VDF | Vinylidene fluoride |
| VTEO | Vinyltriethoxysilane |
References
- D. Xiong, G. Liu, L. Hong and E. J. S. Duncan, Chem. Mater., 2011, 23, 4357–4366 CrossRef CAS
.
- L. Cheng, K. Han, K. Xu, M. R. Gadinski and Q. Wang, Polym. Chem., 2013, 4, 24–36 Search PubMed
- J. Scheirs, Modern Fluoropolymers, Wiley, New York, 1997 Search PubMed
- G. D. Fu, E. T. Kang, K. G. Neoh, C. C. Lin and D. J. Liaw, Macromolecule, 2005, 38, 7593–7600 CrossRef CAS
- J. Furukawa and E. Kobayashi, Rubber Chem. Technol., 1978, 51, 600–646 CrossRef CAS
- G. Hougham, P. E. Cassidy, K. Johns and J. Davidson, Fluoropolymers: Synthesis and Applications, Plenum Publishers, New York, 1999 Search PubMed
- N. K. Sinha and B. Raj, Nucl. Eng. Des., 2010, 240, 2925–2947 CrossRef
- M. Licchelli, M. Malagodi, M. L. Weththimuni and C. Zanchi, Prog. Org. Coat., 2013, 76, 495–503 CrossRef CAS
- I. Wlassics and E. Giannetti, Canadian Patent, 2,182,328, assigned to Ausimont S.p.A, 1997
- B. Ameduri and B. Boutevin, J. Fluorine Chem., 1997, 100, 97–116 CrossRef
- A. L. Logothetis, Perfuoroelastomers and their functionalization, in Macromolecular design of polymeric materials, ed. K. Hatada, T. Kitayama and O. Vogl, New York, Marcel Dekker, 1997, ch. 26, pp. 447–55 Search PubMed
- G. Kostov, B. Ameduri, T. Sergeeva, W. R. Dolbier, R. Winter and G. L. Gard, Macromolecule, 2005, 38, 8316–8326 CrossRef CAS
- D. R. Rexford, US Pat., 3051677, assigned to DuPont de Nemours and Co, Inc., 1964
- J. Wei and J. Qiu, Polymer, 2014, 55, 3818–3824 CrossRef CAS
- C. Fan, B. Li, M. Ren, P. Wu, Y. Liu and T. Chen, RSC Adv., 2015, 5, 18932–18938 RSC
- T. Nakagawa and M. Tatemoto, US Pat., 4158678, assigned to Daikin, 1979
- M. Tatemoto and S. Morita, US Pat., 4361678, assigned to Daikin, 1982
- S. V. Sokolov, V. I. Skobikova, V. I. Purtseladze, T. I. Rabtseva, I. V. Kokotin, N. Y. Chulanova, L. M. Senyushov, M. V. Zhuravlev and A. VanCleeff, US Pat., 5739233, assigned to E. I. DuPont de Nemours and Company, 1998
- G. Tillet, G. Lopez, M. Hung and B. Améduri, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1171–1173 CrossRef CAS
- B. Baradie and M. S. Shoichet, Macromolecules, 2005, 38, 5560–5568 CrossRef
- S. S. Banerjee and A. K. Bhowmick, Polymer, 2013, 54, 6561–6571 CrossRef
- S. S. Banerjee and A. K. Bhowmick, Polymer, 2015, 57, 105–116 CrossRef
- J. Heidarian and A. Hassan, Composites, Part B, 2014, 58, 166–174 CrossRef CAS
- X. Meng, X. Liu, C. Cong and Q. Zhou, Polym. Compos., 2015, 36, 257–263 CrossRef CAS
- D. Lee, J. Hong, C. Lee, J. Oh, S. Kwak and T. Han, Macromol. Res., 2012, 20, 673–681 CrossRef CAS
- Y. Wang, L. Liu, Y. Luo and D. Jia, Polym. Degrad. Stab., 2009, 94, 443–449 CrossRef
- J. S. Furno and E. B. Nauman, Polymer, 1991, 32, 88–94 CrossRef
- N. Hinchiranan, P. Wannako, B. Paosawatyanyong and P. Prasassarakich, Mater. Chem. Phys., 2013, 139, 689–698 CrossRef CAS
- J. Guo, X. Zeng and Q. Luo, J. Elastomers Plast., 2009, 41, 554–573 CrossRef CAS
- J. Guo, X. Zeng, H. Li and Q. Luo, J. Elastomers Plast., 2013, 45, 271–288 CrossRef
- J. Gao, Z. Gu, G. Song, P. Li and W. Liu, Appl. Clay Sci., 2008, 42, 272–275 CrossRef CAS
- D. H. Kim, S. H. Hwang and B. S. Kim, J. Appl. Polym. Sci., 2012, 125, 1625–1635 CrossRef CAS
- A. Hallenbeek and D. Maclachlna, Elastomerics, 1977, 109(l), 41–50 Search PubMed
- M. Gelin and B. Ameduri, J. Fluorine Chem., 2003, 119, 53–58 CrossRef CAS
- C. Boyer, B. Ameduri, B. Boutevin and W. R. Dolbier, Macromolecules, 2008, 41, 1254–1263 CrossRef CAS
- S. Turri, R. Valsecchi, M. Levi, M. Cristini and A. Sanguineti, Eur. Polym. J., 2008, 44, 2951–2961 CrossRef CAS
- B. Boutevin, G. Caporiccio, F. G. Pietrasanta and A. Ratsimihety, Macromol. Chem. Phys., 1998, 199, 61–70 CrossRef CAS
- J. Li, Y. Lu, Y. Li, X. Zhang and S. Qi, Polym.-Plast. Technol. Eng., 2014, 53, 46–53 CrossRef CAS
- J. Legare, E. Thomas, K. Fulford and J. Cargo, Microcontamination'93 Conference Proceedings, Canon Communications, Santa Monica, CA, 1993, pp. 36–46 Search PubMed
- A. Logothetis, Prog. Polym. Sci., 1989, 14, 251–296 CrossRef CAS
- G. Sharama, L. Klintberg and K. Hjort, J. Micromech. Microeng., 2011, 21, 21–25 Search PubMed
- J. Tan, Y. J. Chao, J. W. Van Zee, X. Li, X. Wang and M. Yang, Mater. Sci. Eng., A, 2008, 496, 464–470 CrossRef
- J. Tan, Y. J. Chao, M. Yang, C. T. Williams and J. W. Van Zee, J. Mater. Eng. Perform., 2008, 17, 785–792 CrossRef CAS
- G. Li, J. Tan and J. Gong, J. Power Sources, 2012, 205, 244–251 CrossRef CAS
- R. Souzy and B. Ameduri, Prog. Polym. Sci., 2005, 30, 644–687 CrossRef CAS
- K. K. Jana, A. K. Thakur, V. K. Shahi, D. K. Avasthi, D. Rana and P. Maiti, J. Mater. Chem. A, 2015, 3, 10413–10424 CAS
- V. Gubanov and V. Berenblit, Russ. J. Appl. Chem., 2009, 82, 2242–2247 CrossRef CAS
- B. Ameduri and B. Boutevin, J. Fluorine Chem., 2005, 126, 221–229 CrossRef CAS
- B. Ameduri, B. Boutevin and G. Kostov, Prog. Polym. Sci., 2001, 26, 105–187 CrossRef CAS
- P. Lederer and S. Bowers, Sealing Technology, 2004, 6–9 CrossRef
- J. Li, PhD Dissertation, Beijing University of Chemical Technology, 2013
- M. Zhang and T. P. Russell, Macromolecules, 2006, 39, 3531–3539 CrossRef CAS
- M. Rahimabady, L. Xu, S. Arabnejad, K. Yao, L. Lu and V. P. W. Shim, Appl. Phys. Lett., 2013, 103, 262904 CrossRef
- A. Taguet, B. Ameduri and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2008, 47, 121–136 CrossRef
- B. Ameduri, Macromolecules, 2010, 43, 163–184 Search PubMed
- A. L. Logothetis, Perfluoroelastomers and their functionalization, in Macromolecular design of polymeric materials, ed. K. Hatada, T. Kitayama and O. Vogl, Marcel Dekker, New York, 1997, ch. 26, pp. 447–55 Search PubMed
- E. W. Ross, J. R. Gary and S. Hoover, Liquid fluoroelastomers, Mod. Fluoropolym., 1997, 422–424 Search PubMed
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6686 CrossRef CAS PubMed
- W. Yao, Y. Li and X. Huang, Polymer, 2014, 55, 6197–6211 CrossRef CAS
- F. Boschet and B. Ameduri, Chem. Rev., 2014, 114, 927–980 CrossRef CAS PubMed
- V. A. Petrov, Fluorinated heterocyclic compounds: synthesis, chemistry and applications, Wiley-Vch Verlag GmbH& Co. KGaA, 2009 Search PubMed
- F. Mizuide, H. Tatsu, S. Sokolov, M. Zhuravlev, I. Kokotin and O. Blagodatova, US Pat., 6329471 B1, assigned to Nippon Mektron, Limited, 2001
- M. A. Kader and A. K. Bhowmick, Polym. Degrad. Stab., 2003, 79, 283–295 CrossRef CAS
- S. Mitra, A. Ghanbari-Siahkali, P. Kingshott, K. Almdal, R. H. Kem and A. G. Christensen, Polym. Degrad. Stab., 2004, 83, 195–206 CrossRef CAS
- M. Carenza, S. Lora and G. Pezzin, Radiat. Phys. Chem., 1990, 35, 172–175 CrossRef CAS
- T. Sugama, Mater. Lett., 2001, 50, 66–72 CrossRef CAS
- R. Anilkumar and D. J. Burton, Tetrahedron Lett., 2003, 44, 6661–6664 CrossRef CAS
- Z. Zhang, C. Wang and T. C. Chung, Macromolecules, 2007, 40, 5235–5240 CrossRef CAS
- K. L. Paciorek, L. C. Mitchell and C. T. Lenk, J. Polym. Sci., 1960, 405–413 CrossRef CAS
- W. Schmiegel, Angew. Makromol. Chem., 1979, 77, 39–49 CrossRef
- T. M. Aminabhavi, S. F. Harlapur and O. J. Dale, Polymer, 1997, 38, 2725–2731 CrossRef CAS
- E. Cluff and B. Hundred, US Pat., 3147314, assigned to DuPont de Nemours and Co, Inc., 1964
- F. Mizuide, H. Tatsu, S. V. Sokolov, M. V. Zhuravlev, I. V. Kokotin and O. V. Blagodatova. US Pat., 6174928 B1, assigned to Nippon Mektron, Limited, JP, 2001
- J. Wu, Master Dissertation, Beijing University of Chemical Technology, 2015
- J. Li, Y. Lv, L. Guo, C. Liu, X. Zhang and S. Qi, Acta Polym. Sin., 2013, 1430–1437 CAS
- J. Li, Y. Lv and S. Qi, China Synth. Rubber Ind., 2014, 37, 188–193 CAS
- S. Qi, J. Li, Y. Lu and J. Wang, CN Pat., 103183763 A, assigned to Beijing University of chemical technology, 2013
- S. Qi, J. Li, Y. Lv and J. Wang, CN Pat., 103193919 A, assigned to Beijing University of chemical technology, 2013
- S. Qi, J. Wu, X. Li, Y. Li and X. Zhang, CN Pat., 103980405 A, assigned to Beijing University of chemical technology, 2014
- C. Chen, Y. Liu, D. Wang, W. Tong, X. Zhang and C. Lu, Polym. Mater. Sci. Eng., 2011, 27, 57–59 Search PubMed
- E. M. W. Tsang, Z. Zhang, Z. Shi, T. Soboleva and S. Holdcroft, J. Am. Chem. Soc., 2007, 129, 15106–15107 CrossRef CAS PubMed
- A. Taguet, L. Sauguet, B. Ameduri and B. Boutevin, J. Fluorine Chem., 2007, 128, 619–630 CrossRef CAS
- M. Kanungo, M. Kelly, V. Sambhy, D. Gervast, S. Badesha and C. Chidambarewarapattar, US Pat., 2015/0005454 A1, assigned to Xerox Corporation, 2015
- M. Kanungo, M. Kelly, V. Sambhy, D. Gervast, S. Badesha and C. Chidambarewarapattar, US Pat., 2015/0002581 A1, assigned to Xerox Corporation, 2015
- E. M. W. Tsang, Z. Zhang, A. C. C. Yang, Z. Shi, T. J. Peckham and R. Narimani, Macromolecules, 2009, 42, 9467–9480 CrossRef CAS
- C. Kawashima and T. Yasumura, US Pat., 4472557, assigned to Central Class Company, Limited, 1984
- E. Katoh, K. Kawashima and I. Ando, Polym. J., 1995, 27, 645–650 CrossRef CAS
- R. Souzy, B. Boutevin and B. Ameduri, Macromolecules, 2012, 45, 3145–3160 CrossRef CAS
- A. Taguet, B. Ameduri and B. Boutevin, Fuel Cells, 2006, 6, 331–339 CrossRef CAS
- C. C. Aymes, N. Betz, M. C. Durrier and A. Moel, Nucl. Instrum. Methods Phys. Res., 1999, 151, 377–385 CrossRef
- M. Nasef, H. Saidi, H. Nor, Z. Dahlan and K. Hashim, J. Appl. Polym. Sci., 2000, 37, 2095–2102 Search PubMed
- M. Nasef, H. Saidi, H. Nor and O. Foo, J. Appl. Polym. Sci., 2000, 76, 1–16 CrossRef CAS
- M. Nasef, H. Saidi and H. Nor, J. Appl. Polym. Sci., 2000, 77, 1877–1885 CrossRef CAS
- M. Nasef, H. Saidi and M. Yarmo, J. Appl. Polym. Sci., 2000, 77, 2455–2463 CrossRef CAS
- C. Amyes, N. Yagoubi, N. Betz, A. Moel and D. Ferrier, Chromatographia, 2000, 51, 269–376 Search PubMed
- B. Soresi, Solid State Ionics, 2004, 166, 383–389 CrossRef CAS
- Z. Zhang, E. Chalkova, M. Fedkin, C. Wang, S. N. Lvov and S. Komarneni, Macromolecules, 2008, 41, 9130–9139 CrossRef CAS
- Y. W. Kim, J. T. Park, J. H. Koh, D. K. Roh and J. H. Kim, J. Membr. Sci., 2008, 325, 319–325 CrossRef CAS
- Y. W. Kim, J. K. Choi, J. T. Park and J. H. Kim, J. Membr. Sci., 2008, 313, 315–322 CrossRef CAS
- R. Zhou, W. Liu, Y. W. Leong, J. Xu and X. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 16548–16557 CAS
- C. Moorlag, N. Hu and Y. Qi, US Pat., 7955659 B2, assigned to Xerox Corporation, 2011
- C. Moorlag, N. Hu and Y. Qi, US Pat., 7767758 B2, assigned to Xerox Corporation, 2010
- K. Luo, X. Hu, D. Huang and J. Cheng, CN Pat., 102936319 A, assigned to Guangzhou Glorystar Chemical Co. Ltd, 2012
- N. Hu, K. Luo, G. Lai and Z. He, CN Pat., 102936386 A, assigned to Guangzhou Glorystar Chemical Co. Ltd, 2012
- L. Dong, Y. Wang, J. Yang and C. Xiong, China Synth. Rubber Ind., 2009, 3, 223–226 Search PubMed
- J. D. Li, M. S. Xue, W. Ying and Y. Jian, Special. Purpose. Rubber. Products, 2008, vol. 29, pp. 12–15 Search PubMed
- M. Du, H. Du, Z. Zhang, C. Zhang, Y. Cui and Z. Gong, CN Patent, 104530610 A, assigned to No 53 Res Inst of China North Ind Group Corp, 2014
- P. Paglia and W. Schmiegel, US Pat., 20050277740A1, 2005
- J. Guo, X. Zeng and Q. Luo, China Synth. Rubber Ind., 2009, 32, 5–16 Search PubMed
- N. Hu, D. Huang and K. Luo, CN Pat., 102924861 A, assigned to Guangzhou Glorystar Chemical Co. Ltd, 2013
- A. Taguet, B. Amduri and B. Boutevin, J. Polym. Sci., Part A–1: Polym. Chem., 2006, 44, 1855–1868 CrossRef CAS
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed
- Y. L. Qiao, M. S. Islam, X. D. Yin, K. Han, Y. Yan, J. Y. Zhang, Q. Wang, H. J. Ploehn and C. B. Tang, Polymer, 2015, 72, 428–435 CrossRef CAS
- N. M. L. Hansen, K. Jankova and S. Hvilsted, Eur. Polym. J., 2007, 43, 255–293 CrossRef CAS
- L. Rubatat, Z. Shi, O. Diat, S. Holdcroft and B. J. Frisken, Macromolecules, 2006, 39, 720–729 CrossRef CAS
- Y. Chen, L. Ying, W. H. Yu, E. T. Kang and K. G. Neoh, Macromolecules, 2003, 36, 9451–9461 CrossRef CAS
- T. R. Dargaville, G. A. George, D. J. T. Hill and A. K. Whittaker, Prog. Polym. Sci., 2003, 28, 1355–1376 CrossRef CAS
- C. Tournut, Macromol. Symp., 1994, 82, 99–109 CrossRef CAS
- J. S. Forsythe and D. J. T. Hill, Prog. Polym. Sci., 2000, 25, 101–136 CrossRef CAS
- Y. Urushihara and T. Nishino, Polymer, 2009, 50, 3245–3249 CrossRef CAS
- J. Gao, X. Xu, C. Fan, X. Wang, Y. Dai and X. Liu, Mater. Lett., 2014, 121, 219–222 CrossRef CAS
- I. Banik, S. K. Dutta, T. K. Chaki and A. K. Bhowmick, Polymer, 1999, 40, 447–458 CrossRef CAS
- C. Giovedi, E. S. Pino, M. R. Rossi and L. D. B. Machado, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 2007, 265, 256–259 CrossRef CAS
- N. O. Brace, J. Fluorine Chem., 1999, 93, 1–25 CrossRef CAS
- N. O. Brace, J. Fluorine Chem., 1999, 96, 101–127 CrossRef CAS
- P. M. Murphy, C. S. Baldwin and R. C. Buck, J. Fluorine Chem., 2012, 138, 3–23 CrossRef CAS
- V. Církva, O. I. Paleta, B. Ameduri and B. Boutevin, J. Fluorine Chem., 1995, 75, 87–92 CrossRef
- A. Manseri, B. Ameduri, B. Boutevin, M. Kotora, M. Hajek and G. Caporiccio, J. Fluorine Chem., 1995, 73, 151–158 CrossRef CAS
- A. Manseri, B. Ameduri, B. Boutevin, R. Chambers, G. Caporiccio and A. Wright, J. Fluorine Chem., 1994, 74, 59–67 CrossRef
- L. Zhang, H. Xu, C. Cheng, S. Zhang, Z. Cai and K. Xia, CN Pat., 01139051.4, assigned to Bona Science and Technology De, 2002
- L. Zhang and B. Zhou, CN Patent, 02111543.5, assigned to Zhonglin Material Technology Co., 2002
- C. Boyer, B. Ameduri and M. H. Hung, Macromolecules, 2010, 43, 3652–3663 CrossRef CAS
- M. Tatemoto and T. Amano, Eur Patent, 77998, assigned to Daikin Kogyo Co., Ltd., 1982
- M. Tatemoto and S. Morita, US Pat., 4361678, assigned to Daikin Kogyo Co., Ltd., 1982
- G. K. Kostov, B. Ameduri, F. Boschet and S. Brandstadter, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3964–3981 CrossRef CAS
- M. H. Hung, B. Ameduri and C. Boyer, US Pat., 0105435, assigned to DuPont Performance Elastomers, 2009
- D. P. Carlson, US Pat., 5284920, assigned to DuPont, 1994
- Y. Funakoshi and J. Miki, US Pat., 6919490, assigned to Daikin Industries, Ltd., 2005
- T. Yagi, T. Tsuda, T. Noguchi, K. Sakaguchi, Y. Tanaka and M. Tatemoto, Eur Patent, 0422644, assigned to Daikin, 1990
- D. Carlson, US Pat., 5284920, assigned to E.I. DuPont de Nemours and Co, 1994
- U. Gayer, T. Schuh, V. Arcella and M. Albano, EP 1,231,239 A1, assigned to Ausimont S.p.A., 2002
- C. Boyer, D. Valade, L. Sauguet, B. Ameduri and B. Boutevin, Macromolecules, 2005, 38, 10353–10362 CrossRef CAS
- H. Sawada, T. Tashima, Y. Nishiyama, M. Kikuchi, Y. Goto and G. Kostov, Macromolecules, 2011, 44, 1114–1124 CrossRef CAS
- M. H. Hung, B. Ameduri, A. Soules and B. Boutevin, US Pat., 0324234, assigned to Dupont Performance Elastomers and CNRS, 2011
- M. H. Hung and B. Ameduri, US Pat., 8138274, assigned to Dupont Performance Elastomers and CNRS, 2012
- G. K. Kostov, M. Holan, B. Ameduri and M. H. Hung, Macromolecules, 2012, 45, 7375–7387 CrossRef CAS
- M. H. Hung, B. Ameduri and G. Kostov, US Pat., 2011/0015359, assigned to Dupont Performance Elastomers, 2011
- K. Li, S. Liang, Y. Lu and Q. Wang, Macromolecules, 2007, 40, 4121–4123 CrossRef CAS
- C. Zhao, M. Guo, Y. Lu and Q. Wang, Macromol. Symp., 2009, 279, 52–58 CrossRef CAS
- Y. Lu, J. Claude, F. L. E. Norena and Q. Wang, J. Phys. Chem. B, 2008, 112, 10411–10416 CrossRef CAS PubMed
- L. R. Saint, A. Manseri, B. Ameduri, B. Lebret and P. Vignane, Macromolecules, 2002, 35, 1524–1536 CrossRef
- K. Xu, C. Chanthad, M. A. Hickner and Q. Wang, J. Mater. Chem., 2010, 20, 6291 RSC
- K. Han, Q. Li, C. Chanthad, M. R. Gadinski, G. Zhang and Q. Wang, Adv. Funct. Mater., 2015, 25, 3505–3513 CrossRef CAS
- C. Chanthad, K. Xu, H. Huang and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4800–4810 CrossRef CAS
- D. Rice, US Pat., 3438953, assigned to 3M, 1969
- D. Rice, US Pat., 3457245, assigned to 3M, 1969
- D. Rice, US Pat., 3461155, assigned to 3M, 1969
- J. Li, P. Khanchaitit, K. Han and Q. Wang, Mater. Chem., 2010, 22, 5350–5357 CrossRef CAS
- K. Xu, K. Li, P. Khanchaitit and Q. Wang, Mater. Chem., 2007, 19, 5937–5945 CrossRef CAS
- V. S. D. Voet, M. Tichelaar, S. Tanase, M. C. Mittelmeijer-Hazeleger, G. ten Brinke and K. Loos, Nanoscale, 2013, 5, 184–192 RSC
- V. S. D. Voet, D. Hermida-Merino, G. ten Brinke and K. Loos, RSC Adv., 2013, 3, 7938–7946 RSC
- V. S. D. Voet, G. ten Brinke and K. Loos, J. Polym. Sci., Part A–1: Polym. Chem., 2014, 52, 2861–2877 CrossRef CAS
- V. S. D. Voet, K. Kumar, G. T. Brinke and K. Loos, Macromol. Rapid Commun., 2015, 36, 1756–1760 CrossRef CAS PubMed
- T. C. Chung and A. Petchsuk, Macromolecules, 2002, 35, 7678–7684 CrossRef CAS
- T. Chung and A. Petchsuk, US Pat., 6355749, assigned to Dai-Act, 2002
- M. Chung, Z. Zhang, E. Chalkova, C. Wang, M. Fedkin and S. Komarneni, Proceedings of 212th Meeting of the Electr Chem, 2007 Search PubMed
- Z. Zhang, Z. Wang and T. C. Chung, Macromolecules, 2007, 40, 5235–5240 CrossRef CAS
- R. Severac, PhD Dissertation, University of Montpellier, 2003
- L. Sauguet, PhD Dissertation, University of Montpellier, 2005
- F. Boschet, J. Cracowski, V. Montembault and B. Ameduri, Macromolecules, 2010, 43, 4879–4888 CrossRef CAS
- Y. Patil, A. Alaaeddine, T. Ono and B. Ameduri, Macromolecules, 2013, 46, 3092–3106 CrossRef CAS
- P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS PubMed
- S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57–93 CrossRef CAS
- T. Saito, H. D. Moore and M. A. Hickner, Macromolecules, 2010, 43, 599–601 CrossRef CAS
- L. Sauguet, B. Ameduri and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1814–1834 CrossRef CAS
- R. P. Nielsen, R. Valsecchi, M. Strandgaard and M. Maschietti, J. Supercrit. Fluids, 2015, 101, 124–130 CrossRef CAS
- D. Molina, G. Griffini, M. Levi and S. Turri, Polym. Adv. Technol., 2014, 25, 1082–1088 CrossRef CAS
- A. Soules, B. Ameduri, B. Boutevin and G. Calleja, Macromolecules, 2010, 43, 4489–4499 CrossRef CAS
- H. Kobayashi, Eur Patent, 0665270, assigned to Dow Corning Torray, 1995
- M. T. Maxson, A. W. Norris and M. J. Owen, Fluorosilicones, in Modern fuoropolymers, ed. J. Scheirs, Wiley, New York, 1997, ch. 20, pp. 359–72 Search PubMed
- P. Uritani, Eur Patent, 0967251, assigned to Shin Etsu, 1999
- B. Baradie and M. S. Shoichet, Macromolecules, 2002, 35, 3569–3575 CrossRef CAS
- B. Baradie, M. S. Shoichet, Z. Shen, M. A. McHugh, L. Hong and Y. Wang, Macromolecules, 2004, 37, 7799–7807 CrossRef CAS
- D. Smith and D. Babb, Macromolecules, 1996, 29, 852–860 CrossRef CAS
- R. D. Lousenberg and M. S. Shoichet, Macromolecules, 2000, 33, 1682–1685 CrossRef CAS
- S. Hiroshi and M. Joseph, J. Supercrit. Fluids, 2000, 38, 1139–1145 Search PubMed
- S. Turri, M. Levi, M. Cristini and A. Sanguineti, J. Polym. Res., 2007, 14, 141–145 CrossRef CAS
- W. Kallert, Polyurethane Handbook, ed, G. Oertel, Hanser Publishers, Munich, 1985, pp. 433–443 Search PubMed
- G. Griffini, R. Suriano and S. Turri, Polym. Eng. Sci., 2012, 52, 2543–2551 CAS
- S. Turri, M. Levi, M. Cristini and A. Sanguineti, Polym. Int., 2005, 54, 698–704 CrossRef CAS
- H. R. Allcock, C. A. Crane, C. T. Morissey, J. M. Nelson, S. D. Reeves, C. H. Honeyman and I. Manners, Macromolecules, 1996, 29, 7740–7746 CrossRef CAS
- H. R. Allcock and R. Prange, Macromolecules, 2001, 34, 6858–6863 CrossRef CAS
- S. S. Badesha, G. J. Heeks, R. M. Ferguson, D. J. Gervasi and A. W. Henry, US Pat., 6678495 B1, assigned to Xerox Corporation, 2004
- M. Khajehpour, S. Sadeghi, Y. A. Zehtab and U. Sundararaj, Polymer, 2014, 55, 6293–6302 CrossRef CAS
- J. Wei, S. Jacob and J. Qiu, Compos. Sci. Technol., 2014, 92, 126–133 CrossRef CAS
- H. Ni, S. Wang, Y. Zhou and J. Chen, Polym.-Plast. Technol. Eng., 2010, 49, 1079–1086 CrossRef CAS
- S. Lakshminarayanan, G. A. Gelves and U. Sundararaj, J. Appl. Polym. Sci., 2011, 124, 5056–5063 Search PubMed
- V. Arcella, M. Albano, G. Chiodini, A. Munitillo and G. Brinati, Canadian Patent, 2068754, assigned to Ausimont S.p.A., 1992
- A. Van Cleeff, Fluoroelastomers, in Modern fluoropolymers, ed. J. Scheirs, Wiley, New York, 1997, ch. 32, pp. 597–614 Search PubMed
- A. Van Cleeff, Kautsch. Gummi Kunstst., 1986, 39, 196–198 CAS
- N. Sinha, B. Raj and R. Mukhopadhyay, Sealing Technology, 2014, 4, 10–15 Search PubMed
- P. Kirochko and J. Kreiner, Surf. Coat. Technol., 2001, 84, 91–168 Search PubMed
| This journal is © The Royal Society of Chemistry 2016 |