
Relevant pH and lipase for in vitro models of gastric digestion†
Laura
Sams
ab,
Julie
Paume
b,
Jacqueline
Giallo
b and
Frédéric
Carrière
*a
aCNRS, Aix Marseille Université, Enzymologie Interfaciale et Physiologie de la Lipolyse UMR7282, 31 Chemin Joseph Aiguier, 13402 Marseille Cedex 20, France. E-mail: carriere@imm.cnrs.fr; Fax: +33 4 91 71 5857; Tel: +33 4 91 16 4134
bGERME S.A., Technopôle Marseille Provence Château-Gombert, ZAC la Baronne, 12 Rue Marc Donadille, 13013 Marseille, France
First published on 21st October 2015
Abstract
The development of in vitro digestion models relies on the availability of in vivo data such as digestive enzyme levels and pH values recorded in the course of meal digestion. The variations of these parameters along the GI tract are important for designing dynamic digestion models but also static models for which the choice of representative conditions of the gastric and intestinal conditions is critical. Simulating gastric digestion with a static model and a single set of parameters is particularly challenging because the variations in pH and enzyme concentration occurring in the stomach are much broader than those occurring in the small intestine. A review of the literature on this topic reveals that most models of gastric digestion use very low pH values that are not representative of the fed conditions. This is illustrated here by showing the variations in gastric pH as a function of meal gastric emptying instead of time. This representation highlights those pH values that are the most relevant for testing meal digestion in the stomach. Gastric lipolysis is still largely ignored or is performed with microbial lipases. In vivo data on gastric lipase and lipolysis have however been collected in humans and dogs during test meals. The biochemical characterization of gastric lipase has shown that this enzyme is rather unique among lipases: (i) stability and activity in the pH range 2 to 7 with an optimum at pH 4–5.4; (ii) high tensioactivity that allows resistance to bile salts and penetration into phospholipid layers covering TAG droplets; (iii) sn-3 stereospecificity for TAG hydrolysis; and (iv) resistance to pepsin. Most of these properties have been known for more than two decades and should provide a rational basis for the replacement of gastric lipase by other lipases when gastric lipase is not available.
 Laura Sams | Laura Sams, born in 1991, obtained the Engineering degree in Food Science from AgroSup Dijon in 2013 after internships at Professor Harry Wichers’ Laboratory of Food and Biobased Research in Wageningen, The Netherlands, and Royal Canin, Aimargues, France. She is currently preparing her PhD on lipases for in vitro digestion models, under the supervision of Frédéric Carrière, and is employed (CIFRE contract) by GERME S.A., Marseille, France. |
 Julie Paume | Julie Paume, born in 1982, has an educational background in Applied Mathematics and Informatics (BSc from Avignon University, 2006) and Natural and Technological Risk Management (MSc from Aix-Marseille University, 2008). She joined GERME S.A. in 2008 and became R&D Director in 2011. Among various duties, she is involved in the development of lipase production and in vitro digestion assays for applications in the food and pharma industries. |
 Jacqueline Giallo | Jacqueline Giallo, born in 1955, has an educational background in Applied and Industrial Microbiology (BSc in 1978 and MSc in 1979). She obtained a PhD in Microbiology at Aix-Marseille University (1984) on the study of cellulolytic bacteria. Since 1981, she has been the Chairman and Chief Executive Officer of GERME S.A., a biotechnology company located in Marseille, France, and dedicated to the production of biomolecules and process development based on microorganisms. She has been involved in various projects dedicated to lipase production, purification and biochemical characterization. She created a collaborative laboratory with EIPL in 2006 to transfer research outcomes into industrial applications and research services. |
 Frédéric Carrière | Frédéric Carrière, born in 1963, is Director of Research at the Centre National de la Recherche Scientifique (CNRS) and Head of Enzymology at the Interfaces and Physiology of Lipolysis (EIPL) laboratory, a joint unit of CNRS and Aix-Marseille University, France. He received a degree in Chemical Engineering from Marseille High School of Chemistry in 1986 and a PhD in Enzymology from Aix-Marseille University in 1992. He has 25 years of expertise in the field of lipids and lipolytic enzymes and has around 180 publications dealing with the structure–function relationships and physiological roles of digestive lipases (visit his website at http://eipl.cnrs-mrs.fr/). |
Introduction
There is growing interest in in vitro digestion models because they are useful tools for studying food bioaccessibility, digestibility and bioavailability without, or before, performing animal and human studies.1 This interest is not limited to food research: in vitro digestion models find important applications in pharmaceutical research for preclinical studies of oral drug bioavailability.2 These models are critical for establishing good in vitro–in vivo correlations when drug dispersion and dissolution are highly dependent on digestive processes, as experienced with poorly water soluble drugs and lipid-based formulations for instance.3The development of in vitro digestion models relies on the availability of in vivo data such as digestive enzyme secretion and levels, pH values and their dynamic variations along the GI tract. A first level of confidence in the available data lies in their mode of collection. It is very important for instance that enzyme output/secretions have been measured in the course of meal digestion because meal components have a stabilizing effect: by providing their respective targets (lipids, proteins, carbohydrates) to digestive enzymes (lipases, proteases, amylases), they limit the probability of autolysis by proteases; by buffering gastric contents, these components also prevent enzyme denaturation at low pH and further proteolysis by pepsin. These processes have been well described in the case of gastric lipase which is rapidly degraded under fasting conditions or pharmacological stimulation of its secretion.4,5 Highly variable concentrations of gastric lipase in fasting human gastric juice have thus been reported.6 Degradation of pancreatic enzymes is even more pronounced7 and researchers involved in the purification and characterization of pancreatic enzymes from pancreatic juice and pancreas have always taken great care to avoid protease activation leading to autolysis and poor stability of these sources of enzymes.8,9 Storage conditions of samples before analysis are also critical. Proteolysis is often promoted upon the freezing/thawing process in the absence of protease inhibitors or other agents, protecting enzymes from degradation such as bile salts and meal components.10,11 Once these potential artifacts have been discarded, relevant data can be extracted from the literature and used for designing both dynamic and static digestion models, the latter being the most widely used ones because of their simplicity, lower cost and higher throughput.1
A major difficulty with static models is however the selection of a single set of parameters (pH, enzyme concentration, meal dilution, mixing) supposed to be representative of digestion at a given location within the GI tract. This difficulty is mainly encountered in intragastric digestion where variations in pH and enzyme concentration are much broader than those occurring in the small intestine.12–14 Reproducing the two main functions of the stomach is also challenging: its higher part, the fundus, has a major contribution to gastric acid and digestive enzyme (pepsin, gastric lipase) secretion, whereas the lower part, the antrum, generates mechanical forces to mix, disrupt and transport gastric content by peristaltic movements.15–17 This has led to the development of many intragastric digestion models with different parameter sets.1,18–24 We will not discuss here the attempts to reproduce gastric mixing and fluid flow and the use of pepsin, which have been covered in previous articles and reviews,16,25–27 and we will focus on the choice of pH and lipase for mimicking gastric conditions. In a majority of gastric digestion models, low acidic pH values (1.4–3) are used but their relevance for meal digestion is questionable. Gastric lipase, the first enzyme of the GI tract involved in fat digestion,14,28 is often replaced by other lipases of microbial origin without demonstration of their equivalence. The biochemical properties and specificity of gastric lipase, such as sn-3 stereospecificity for triglyceride (TAG) hydrolysis,29–35 are however unique among known lipases. These various points will be illustrated based on data collected in humans and dogs during test meals. The aim of this review is to provide reliable data on gastric pH and lipase to be used for designing in vitro digestion models.
Overview of in vitro digestion models including a gastric phase
A literature survey by Web of Science™ shows an exponential increase in the number of publications with the “in vitro digestion model” as the topic over the last few years. Among these publications (>6000), we selected and analyzed 340 articles giving detailed data on the type of model used (static vs. dynamic), the number of phases involved (oral, gastric, duodenal/small intestinal, colonic) and key parameters like pH values and the source of digestive enzymes. Static models are the most predominant (95%) and a gastric phase is present in 84% of these models.The pH values and the source of lipase, if any, used in static models and gastric phase are highly variable (Table 1). The pH of the gastric phase varies between 1 and 5.5 and in vitro gastric digestion assays can be classified into three groups: pH < 2 (21%), pH 2 (53%) and pH > 2 (26%). The majority of in vitro gastric digestion experiments are therefore performed at a very low pH supposed to reflect gastric conditions.
| Digestion phase | Parameter | Value/origin | % of total in vitro models |
|---|---|---|---|
| Gastric phase | pH | <2 | 21 |
| 2 | 53 | ||
| >2 | 26 | ||
| Lipase | No lipase | 94.9 | |
| Human gastric juice | 2.3 | ||
| Rhizopus oryzae lipase | 0.9 | ||
| Aspergillus niger lipase | 0.45 | ||
| Purified gastric lipase | 1 | ||
| Unknown origin | 0.45 | ||
| Duodenal phase | pH | <6.5 | 12 |
| 6.5–7.5 | 80 | ||
| >7.5 | 8 | ||
| Lipase | No lipase | 9.6 | |
| Human duodenal juice | 4.3 | ||
| Pancreatin | 70.9 | ||
| Lipase from hog | 10.4 | ||
| Pancreatin + lipase from hog | 3.9 | ||
| Phospholipase A2 | 0.4 | ||
| Cholesterol esterase | 0.4 |
Contrary to pH and pepsin, the use of a lipase during the gastric phase is clearly neglected in most publications. Indeed, only 5% of publications on static digestion models use a lipase to take into account intragastric lipolysis, against 39% with dynamic digestion models. Different sources of lipases can be identified but most published studies use human gastric juice and commercially available microbial lipases from Rhizopus oryzae and Aspergillus niger. Lipases from Candida rugosa, Pseudomonas fluorescens, Rhizomucor miehei36 and Burkholderia cepacia37 were also used in a few studies. The use of gastric lipase purified from human gastric juice or other sources (animal tissue extract, recombinant lipase) is still limited to laboratories producing this enzyme. Lipase concentrations/activities are highly variable and depend on the source of lipase. With gastric lipase, the choice of lipase concentration/activity is usually based on in vivo data,38,39 the mean levels found in human gastric juice (around 100–110 μg mL−1, which is equivalent to 120–130 U mL−1 using tributyrin as the reference substrate5,28) and the meal to gastric juice volume ratio. For instance, a concentration of 17 μg mL−1 gastric lipase is chosen to simulate the gastric conditions at 50% gastric emptying when gastric juice is diluted 6-fold by the meal.38 In the case of microbial lipase, some digestion assays are performed with 3300 μg mL−1 of Aspergillus niger lipase with a specific activity of around 12 U mg−1 (i.e., 39 U mL−1) while some other assays are performed with 200–250 μg mL−1 of Rhizopus oryzae lipase with a specific activity of 80 U mg−1 (i.e., 20 U mL−1).40–43 Variable levels of the same lipase can also be found.40–42,44,45 For instance, the mass concentration of Rhizopus oryzae lipase varies from 200 μg mL−1 to 2700 μg mL−1 (10-fold), while the activity level varies from 17.14 U mL−1 to 110 U mL−1 (6.5-fold).42–45 It is therefore difficult to compare these digestion assays supposed to mimic gastric digestion. It is even more difficult to find a rational basis for the choice of lipase mass concentration and activity in the absence of any publication comparing these microbial lipases with gastric lipase and gastric juice under in vitro digestion conditions. The definition of lipase units (U) is also a major concern because the activities of the lipase sources are often measured using specific assay conditions for each lipase and units cannot be compared.
Variations in gastric pH during meal digestion
The pH of gastric contents and its variations during a meal have been measured on various occasions in healthy volunteers from samples collected by using naso-gastric tubes.13,28,46–48 Gastric pH is not a constant parameter and its value changes continuously during digestion. After meal ingestion, gastric pH increases from 1.0–1.5 (basal fasting conditions) to 5–7 depending on the type of meal and its buffering capacity. It then decreases due to meal dilution by gastric acid secretion before returning to basal conditions after around 3 hours. Gastric emptying also contributes to this pH decrease by removing the meal components from the gastric contents and reducing their buffering effects. These pH variations with time are shown in Fig. 1A and C for liquid and solid–liquid test meals, respectively. Large inter-individual variations are usually observed that result in a great dispersion of pH values plotted as a function of time. With a solid–liquid meal for instance, the pH of gastric contents at 60 min after meal ingestion can range between 6 and 1.5 (Fig. 1C). It is therefore very difficult to define the gastric pH after 1 hour of digestion. The main cause of this variability is gastric emptying and the residence time of the meal in the stomach that varies with the type of food ingested. The rate of gastric emptying is known to be one of the main factors of the variability of postprandial events,49,50 including pH variations, with significant inter- and intra-individual variations in healthy volunteers.51 When meal gastric emptying is measured using a non-absorbable marker like PEG 4000, the gastric pH variations can be plotted as a function of gastric emptying instead of time (Fig. 1B and D). This non-conventional representation reduces the inter-individual variability and gives a new picture of the pH variations in gastric contents during meal digestion.12,28 It thus appears that gastric pH remains rather high and decreases at a slow rate until 50–60% of the meal is emptied from the stomach (Fig. 1B and D). With a liquid test meal, the gastric pH is still found between pH 4 and 7 at 50% gastric emptying with a mean value of 5.5 (Fig. 1B). With a solid meal, the mean gastric pH at 50% gastric emptying is 4.6 (Fig. 1D). It is worth noting that these values are found within the pH range [4–5.4] of optimum activity of gastric lipase on long chain triglycerides.52–54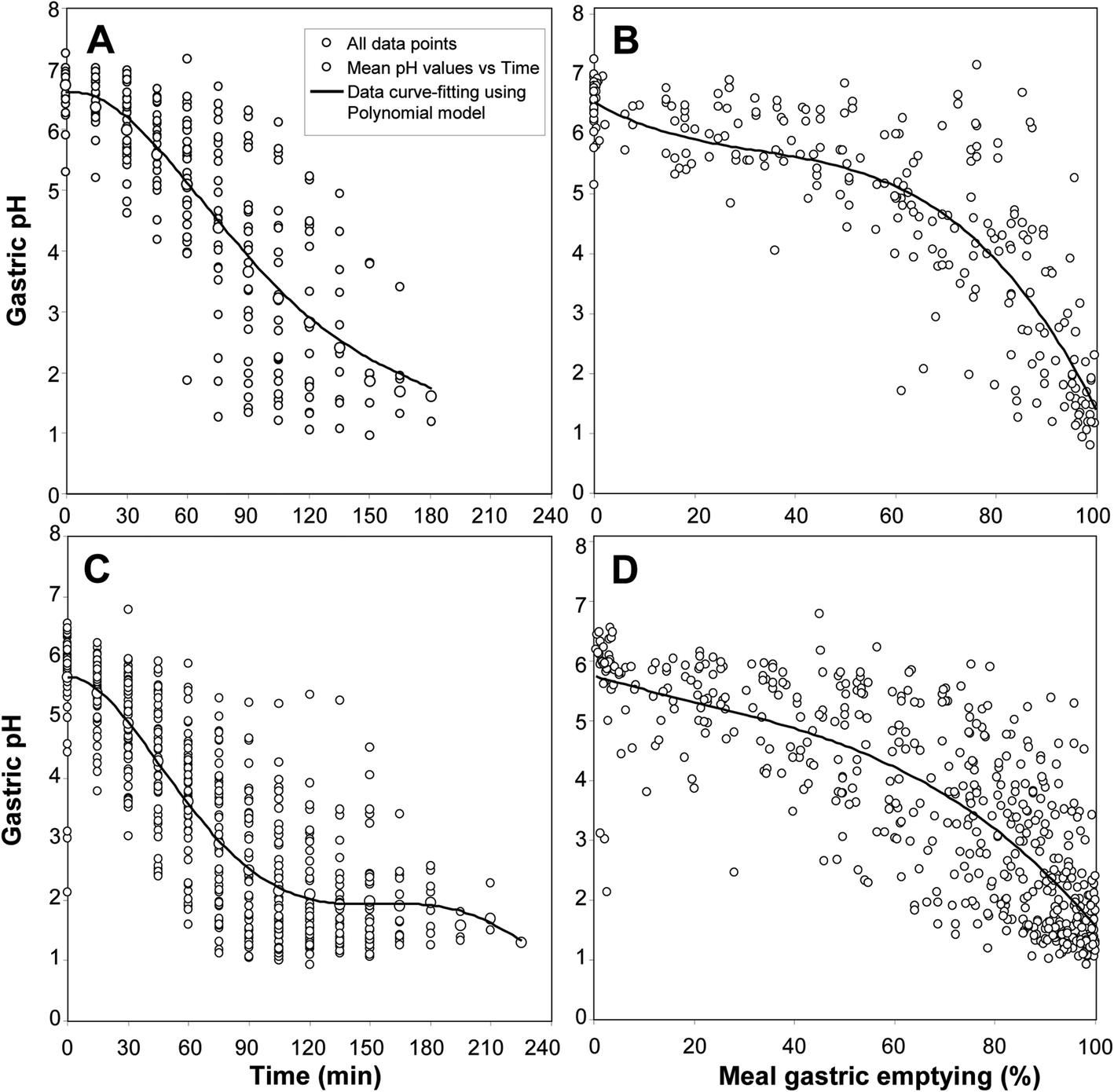 | ||
| Fig. 1 Variations in gastric pH during test meal digestion in healthy volunteers. Panel A: pH variation as a function of time during a liquid test meal (256 values from 30 individual experiments28,48,63). Curve-fitting equation obtained by polynomial regression: y = 6.5905 + 0.0061x − 0.0008x2 + 6 × 10−6x3 − 10−8x4, R2 = 0.6456; panel B: the same pH values plotted as a function of meal gastric emptying (%). The curve-fitting equation obtained by polynomial regression: y = 6.6731 − 0.0484x + 0.0011x2 − 10−5x3, R2 = 0.7412; panel C: pH variation as a function of time during a solid–liquid test meal (545 values from 53 individual experiments13,63). The curve-fitting equation obtained by polynomial regression: y = 5.672 + 0.002x − 0.0013x2 + 10−5x3 − 5 × 10−8x4 + 7 × 10−11x5, R2 = 0.6923; panel D: the same pH values plotted as a function of meal gastric emptying (%). The curve-fitting equation obtained by polynomial regression: y = 5.7531 − 0.0245x + 0.0002x2 − 4 × 10−6x3, R2 = 0.6506. | ||
If we consider the pH value used for the gastric phase in 55% of static in vitro digestion models, namely pH 2, this pH value corresponds to 95% of meal gastric emptying (Fig. 1B and D). This pH is therefore close to that of fasting conditions and is not representative of conditions where most of the meal is still present inside the stomach.
Variations in gastric lipase levels during meal digestion
Gastric enzyme concentrations are also affected by the rate of gastric emptying. Many investigations have been performed to determine the relationship between gastric emptying and regulatory processes induced by hormones or neurotransmitters like enzyme secretions,55–57 as well as the physical properties of test meals.58–62 These correlations have usually been established on a time basis. In the case of human gastric lipase (HGL), however, the enzyme levels in gastric contents can be expressed both as a function of time (Fig. 2A and C) and gastric emptying (Fig. 2B and D). As previously shown with gastric pH (Fig. 1), plotting the HGL concentration as a function of gastric emptying instead of time allows reducing the inter-individual variability. After ingestion of the test meal, the HGL concentration in the gastric content is low because of the high dilution of basal gastric juice by the meal. Then the level of secretion increases because of the stimulation by the meal and at 50% gastric emptying the HGL concentration is around 15–20 μg mL−1. A drastic increase in the HGL concentration is observed after 70–80% gastric emptying. When meal gastric emptying is completed, the lipase concentration reaches the level found under basal conditions, i.e. in pure gastric juice. The mean concentration of HGL in gastric juice is 108 ± 52 μg mL−1, which corresponds to 130 ± 62 U ml−1 when HGL activity is measured with tributyrin as the substrate and shows a specific activity of 1200 U per mg of enzyme.5 The total amount of HGL secreted during a meal is around 20 mg.13,28,48,63 Like gastric pH, HGL concentration undergoes large variations during a meal and it is not obvious to select a representative value to simulate the intragastric process of lipolysis with in vitro static models. In the in vitro digestion studies with a static model and using gastric lipase, an enzyme concentration corresponding to 50% gastric emptying (16–17 μg mL−1) was usually chosen,38,63–68 together with a pH value of 5.5. This pH value corresponds to the mean pH of gastric contents at 50% gastric emptying (liquid meal) and is also close to the pH (5.4) at which the optimum activity of HGL is measured using Intralipid, a soybean oil emulsion.52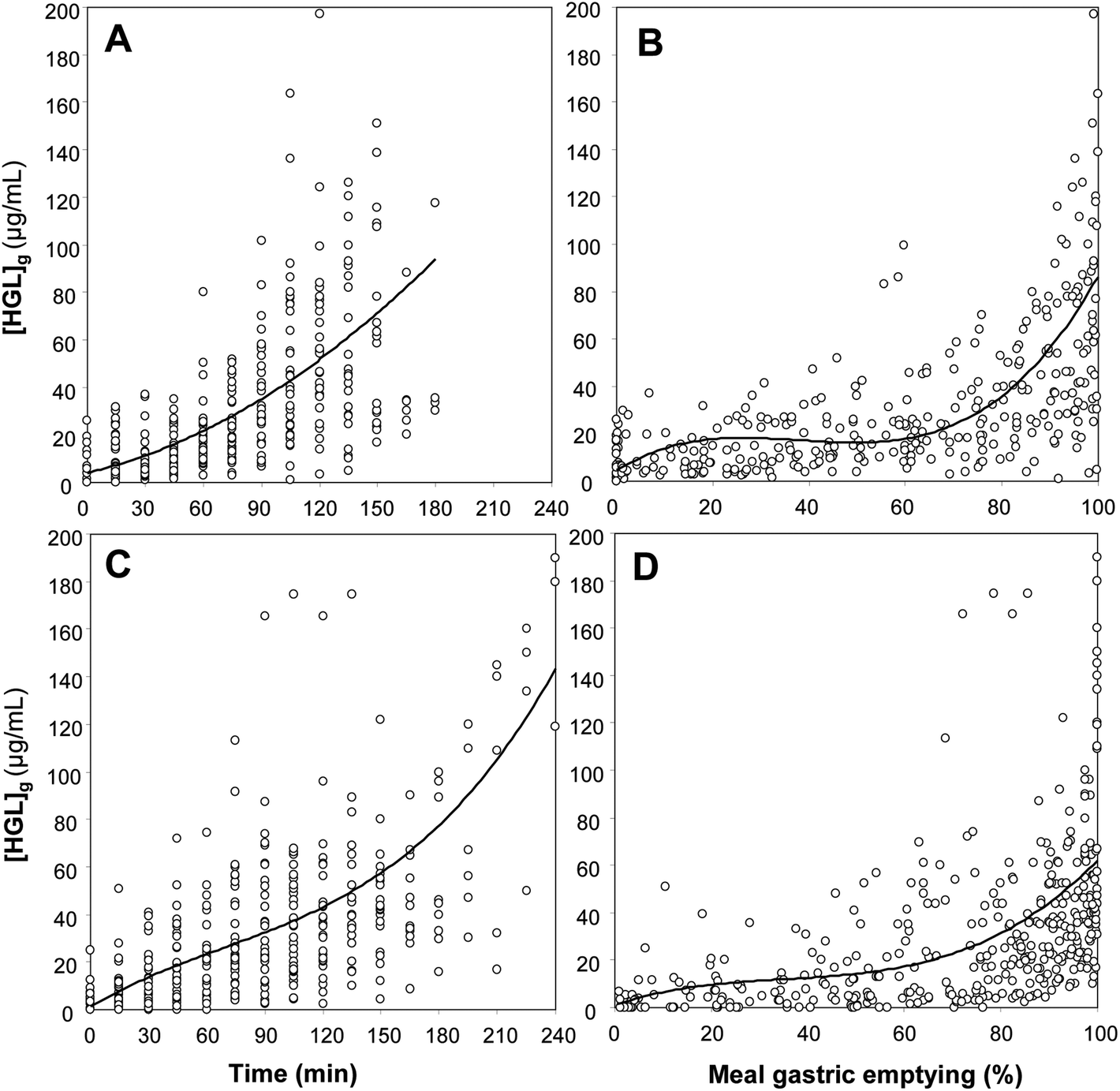 | ||
| Fig. 2 Variation of the gastric lipase concentration in gastric contents ([HGL]g) during test meal digestion in healthy volunteers. Panel A: [HGL]g variation as a function of time during a liquid test meal (256 values from 30 individual experiments28,48,63). The curve-fitting equation obtained by polynomial regression: y = 4.041 + 0.1944x + 0.0017x2, R2 = 0.471; panel B: the same [HGL]g values plotted as a function of meal gastric emptying (%). The curve-fitting equation obtained by polynomial regression: y = 4.6117 + 1.2653x − 0.0368x2 + 0.0003x3, R2 = 0.5459; panel C: [HGL]g variation as a function of time during a solid–liquid test meal (545 values from 53 individual experiments13,63). The curve-fitting equation obtained by polynomial regression: y = 1.2388 + 0.485x − 0.0027x2 + 10−5x3, R2 = 0.3307; panel D: the same [HGL]g values plotted as a function of meal gastric emptying (%). The curve-fitting equation obtained by polynomial regression: y = 0.9692 + 0.7024x − 0.0165x2 + 0.0002x3, R2 = 0.1918. | ||
Unique biochemical properties of gastric lipase
Gastric lipase secretion, like those of gastric acid and pepsinogen, is stimulated by the gastrointestinal hormone gastrin,5,69–71 but other signals such as stomach motion, cholinergic stimuli4 and test meals28 also trigger gastric lipase secretion.72–75 As a result, an appreciable digestion of fat by HGL occurs in the stomach where 10 to 25% of dietary TAG acyl chains can be released.13,28,63 This significant contribution to fat digestion in the gastric environment is due to the specific properties of HGL that can be considered as an extremophilic enzyme.76HGL is a triacylglycerol hydrolase (EC 3.1.1.3) consisting of a 379-amino acid polypeptide of 43 kDa77 with an α/β hydrolase fold.78 Native HGL purified from human gastric juice is highly N-glycosylated (15% w/w), which results in a global molecular mass of 50 kDa and the existence of four major isoforms with isoelectric point ranging from 6.8 to 7.4.79 Like other lipases, it is a serine hydrolase with a catalytic triad (Ser153-His353-Asp324) and an oxyanion hole (Gln154, Leu67), and the access to its active site is controlled by an amphiphilic lid domain.78,80,81 Whereas the first X-ray structure of HGL (Protein Databank ID: 1HGL) was obtained with the lid in the closed conformation78 (Fig. 3A and C), the crystallization of recombinant dog gastric lipase (rDGL) covalently inhibited by a phosphonate inhibitor allowed one to solve a second 3D structure of a gastric lipase with the lid in the open conformation (Protein Databank ID: 1K8Q).80 The conformational changes occurring upon lid opening in HGL could be deduced from these two structures since DGL82 and HGL77 share 86% amino acid sequence identity (Fig. 3B and D). Fig. 3 shows how the opening of the amphiphilic lid gives access to the active site. While lid polar residues are exposed in the closed conformation, hydrophobic residues become exposed upon lid opening and form a large hydrophobic ring surrounding the active site entrance and constituting the interfacial recognition site (IRS).
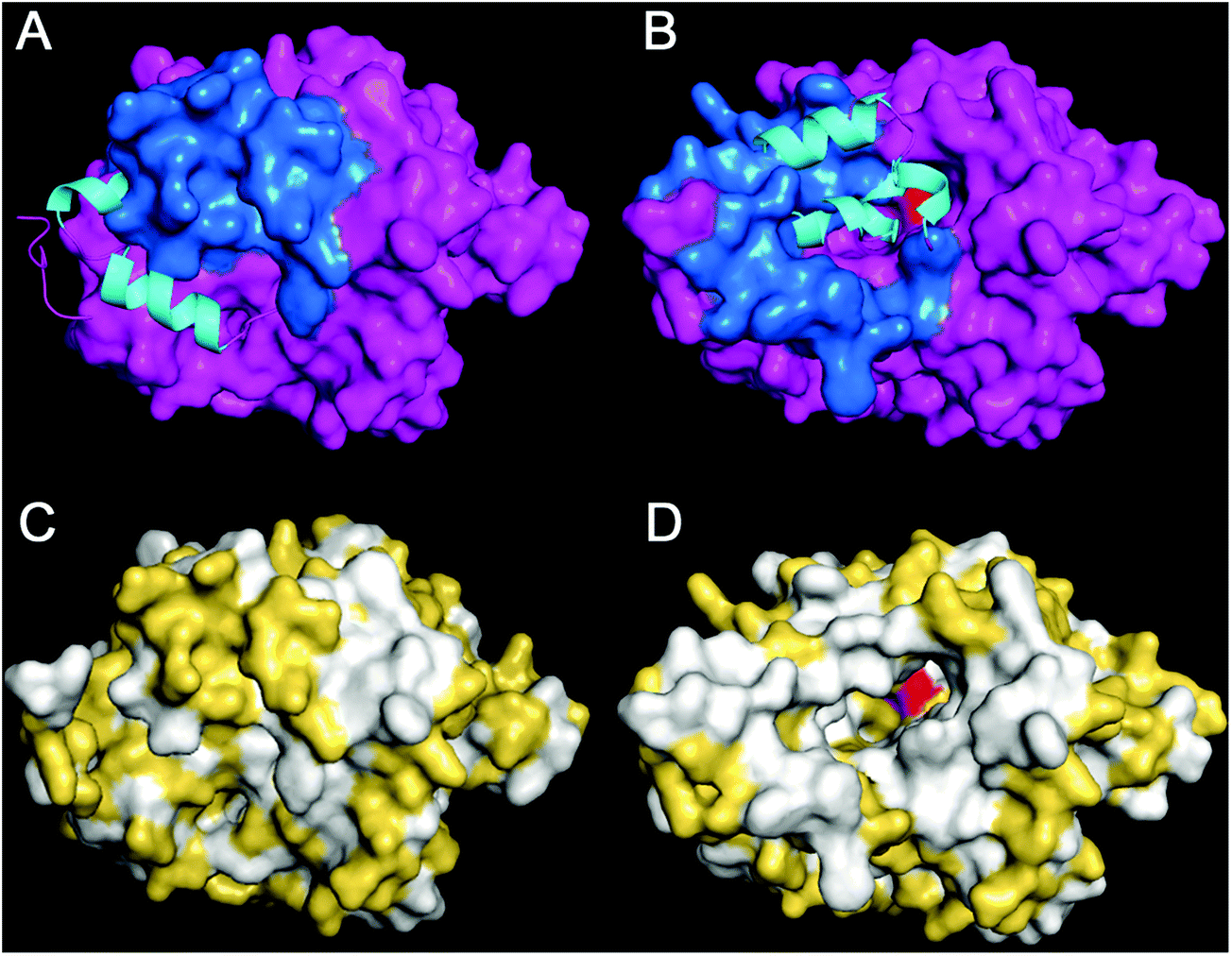 | ||
| Fig. 3 Structure of human gastric lipase. Panel A: Molecular surface representation of the HGL 3D structure with the lid in the closed conformation obtained by X-ray crystallography (Protein Databank ID: 1HGL).78 All amino acid residues are shown in purple, except the lid colored in blue. The N-terminal end (1–9) of HGL is not defined in the electronic density and is only shown as Cα-tracing on the left side of HGL. The conformation of the open lid is shown as a ribbon model with α-helices in cyan. Panel B: 3D model of HGL with the lid in the open conformation, built from HGL-rDGL sequence alignment and the known X-ray structure of open rDGL (Protein Databank ID: 1K8Q).80 The conformation of the closed lid is shown as a ribbon model with α-helices in cyan. The active site serine residue (Ser153) is shown in red. Panels C and D: the same views as A and B, respectively, but hydrophobic amino acids are colored in white and polar amino acids in yellow. | ||
HGL is highly stable under acidic conditions and in gastric juice at pH values ranging from 2 to 7, especially between pH 3 and 5 with half-inactivation times >24 hours.5 Its stability decreases below pH 2 and above pH 7 with half-inactivation times of 43 ± 9 min at pH 1 and 24 ± 18 min at pH 8.5 HGL shows a maximum activity at pH 5–5.4 on long chain TAG,52 whereas most lipases show optimum activity at pH 7 or above. This particular property of gastric lipase does not fit with the known mechanism of action of serine hydrolases with a catalytic triad and a histidine residue involved in the charge relay system.83 Indeed, the nucleophilic character of the serine residue depends on the ionization of histidine and is normally enhanced at pH values above the histidine pKa (6.5). The 3D structures of HGL and rDGL did not reveal any specific features or charged residues in the vicinity of the catalytic triad that could suggest a lower pKa for the catalytic histidine residue His353. Moreover, it was shown that gastric lipase acts on a soluble substrate (vinyl butyrate) with an optimum activity above pH 7, which suggests that gastric lipase is able to hydrolyze ester bonds via the classical mechanism of serine hydrolases.84 The optimum activity of gastric lipase is however shifted towards lower pH values when the concentration of vinyl butyrate exceeds the solubility limit and an oil-in-water phase appears.84 Under these conditions, gastric lipase must first bind at the lipid–water interface before the insoluble substrate is hydrolyzed, which suggests that the lipase adsorption preferentially occurs at low pH. Experiments performed with long chain TAG emulsions confirmed that gastric lipase binds optimally to the oil–water interface at low pH values.84 To study the effects of the pH on the adsorption step independently from substrate hydrolysis, gastric lipase adsorption on solid hydrophobic surfaces was monitored by total internal reflection fluorescence, as well as using a quartz crystal microbalance. Both techniques showed a pH-dependent and reversible gastric lipase adsorption process, which was optimal at pH 5.84 Similar results were obtained when the pH-dependent adsorption of gastric lipase at the lipid–water interface was studied with phospholipid monomolecular films.85 The optimum activity of gastric lipase at acidic pH can therefore be explained by a better adsorption of the enzyme at the lipid–water interface at low pH and the fact that the lipase adsorption is the rate limiting step in the overall process of lipolysis.84
In the stomach, HGL mainly converts TAGs into diglycerides (DAG) and free fatty acids (FFA)28 but in vitro, gastric lipase is able to cleave the three ester bonds of TAGs under conditions optimized to reach high hydrolysis levels.53 The limited action of HGL on gastric contents is due to its inhibition by the lipolysis reaction products, long chain FFAs, that accumulate at the lipid–water interface in the absence of acceptors,52,86 and also to the decrease in gastric pH below optimum values for HGL activity. Once it is emptied in the duodenum with the chyme, HGL finds more favourable conditions for its activity and can still contribute to fat digestion as shown in chronic pancreatitis patients with no pancreatic lipase.13
HGL shows a high activity on TAG and DAG substrates forming oil-in-water emulsions, and much lower activity on monoglycerides (MAG).87,88 Gastric lipase can also hydrolyze mono- and di-esters of polyethyleneglycols (PEG) with various chain lengths,87,88 but has no activity on phospholipids and cholesterol esters.53 HGL shows its highest activity on tributyrin and this short chain substrate has been used for developing a sensitive standard assay of gastric lipase.28,52 Nevertheless, the rates of short, medium and long acyl chain TAG hydrolysis by gastric lipase are found to be in the same order of magnitude.52,53 Whatever the substrate, the optimum activity of gastric lipase is always measured in the pH range 4–6,52,53,87–89 except when the substrate is partly soluble like vinyl butyrate and is used at a concentration below the solubility limit.84
Another specificity of HGL is the fact that this lipase is active in the presence of physiological (micellar) concentration of bile salts52,90 whereas most lipases are inhibited and human pancreatic lipase (HPL) requires a specific protein cofactor to display its activity under these conditions91 (Fig. 4A). Bile salts are strong surfactants that compete with lipases for adsorption at the lipid–water interface and thus can prevent the enzyme from reaching its substrate. HPL counteracts these effects by forming a stoichiometric complex with colipase that anchors the lipase–colipase complex at the interface.91,92 HGL is a more tensioactive enzyme than HPL and does not require a cofactor for adsorption at the lipid–water interface in the presence of micellar concentrations of bile salts,74 probably because of its amphiphilic structure (Fig. 3). The high tensioactivity of gastric lipase has been shown by adsorption studies onto monomolecular films (Fig. 4B). The critical surface pressure for HGL penetration into a monolayer of egg phosphatidylcholine (PC) spread at the air–water interface is 23–25 mN m−1 (ref. 93 and 94) whereas that of HPL is 15–18 mN m−1.91,93 HGL is thus as tensioactive as the HPL–colipase complex (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol/mol) that penetrates into the egg PC monolayer with a critical surface pressure of insertion of 27 mN m−1.93 This property of HGL is important for its biological function: HGL can act in the duodenum where it was found to be active and stable;13,28 HGL can initiate the lipolysis of TAG emulsions covered by phospholipids, which are resistant to pancreatic lipase.94 Gastric lipase thus plays an essential role in the lipolysis of milk fat globules39,95 and triggers the activity of pancreatic lipase.94,96
:1 mol/mol) that penetrates into the egg PC monolayer with a critical surface pressure of insertion of 27 mN m−1.93 This property of HGL is important for its biological function: HGL can act in the duodenum where it was found to be active and stable;13,28 HGL can initiate the lipolysis of TAG emulsions covered by phospholipids, which are resistant to pancreatic lipase.94 Gastric lipase thus plays an essential role in the lipolysis of milk fat globules39,95 and triggers the activity of pancreatic lipase.94,96
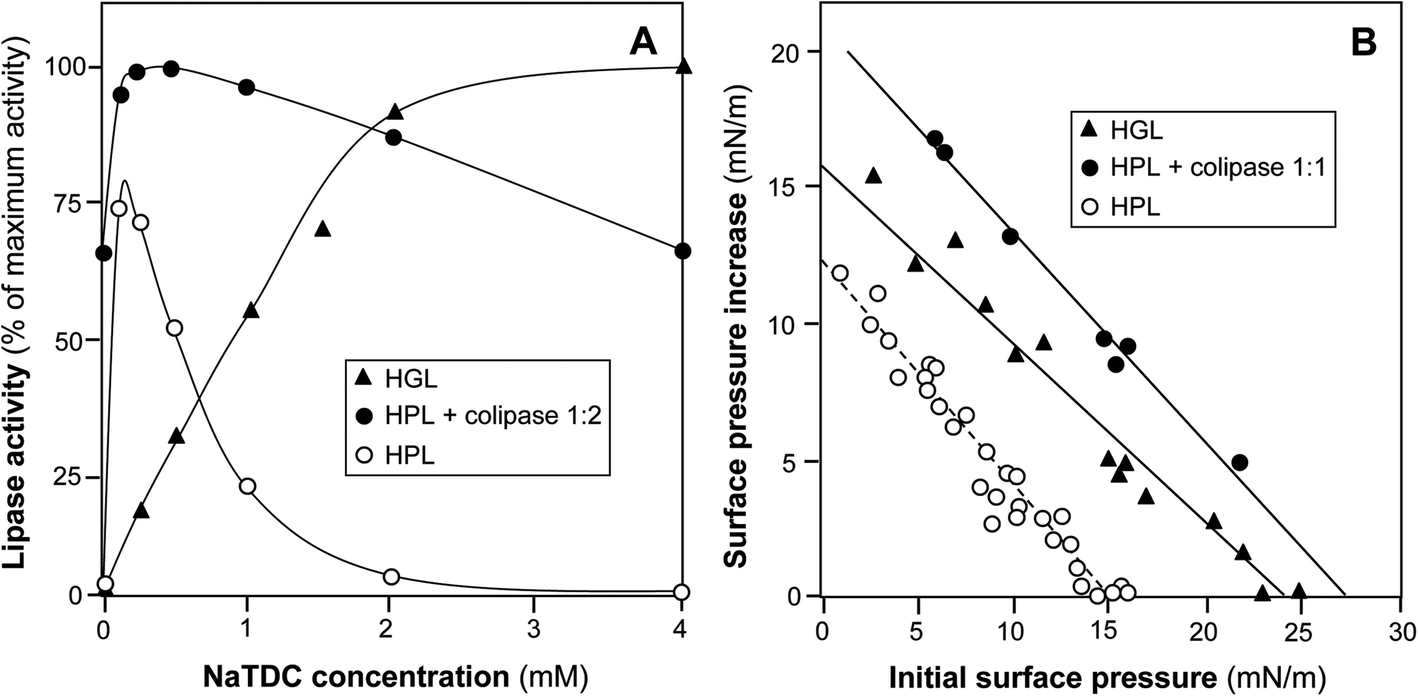 | ||
| Fig. 4 Comparison of HGL and HPL interfacial properties. Panel A: effects of bile salts (NaTDC, sodium taurodeoxycholate) on the lipase activity of HGL, HPL and HPL–colipase complex (2-fold molar excess of colipase); adapted from ref. 52, 74 and 155. Panel B: adsorption/penetration of HGL, HPL and HPL–colipase complex (1:1 molar ratio) onto the egg phosphatidylcholine monolayer spread at the air–water interface. Data for HGL are from ref. 93 and 145: Data for HPL and HPL–colipase complex are from ref. 91. | ||
Although gastric lipase can hydrolyze the three ester bonds of TAG molecules, this enzyme has however a preference for hydrolyzing the ester bond at the sn-3 position of TAGs.29–33,35,97 This stereopreference of gastric lipase has been demonstrated using synthetic chiral TAG,97 enantiomeric glyceride analogs31 and prochiral triglycerides like triolein.30 In the latter case, the sn-3 stereopreference of gastric lipase gives rise to a transient enantiomeric excess of 1,2-sn-diolein in the course of TAG hydrolysis (Fig. 5A),33 that can be used as a tracer of gastric lipase activity in vivo.32 Indeed, a large enantiomeric excess (35–65%) of 1,2-sn-diolein was observed in gastric contents of dogs which received a test meal containing triolein as the sole source of fat (Fig. 5B).32 The enantiomeric excess of 1,2-sn-diolein is only transient and decreases with the hydrolysis level because 1,2-sn-diolein is further hydrolyzed and converted into 2-sn-monoolein (Fig. 5A). The relative specificity constants of TAG conversion into 1,2-sn-DAG (or 2,3-sn-DAG) and 1,2-sn-DAG (or 2,3-sn-DAG) conversion into 2-sn-MAG have been determined for rDGL and some other lipases (Table 4). These specificity constants allow the quantitative comparison of the stereoselectivity fingerprints for various lipases acting on the prochiral triolein substrate.34 While rDGL preferentially forms 1,2-sn-DAG from TAG and then preferentially hydrolyzes 1,2-sn-DAG versus 2,3-sn-DAG, the lipase from Rhizomucor miehei shows a reverse stereospecificity (Table 4).
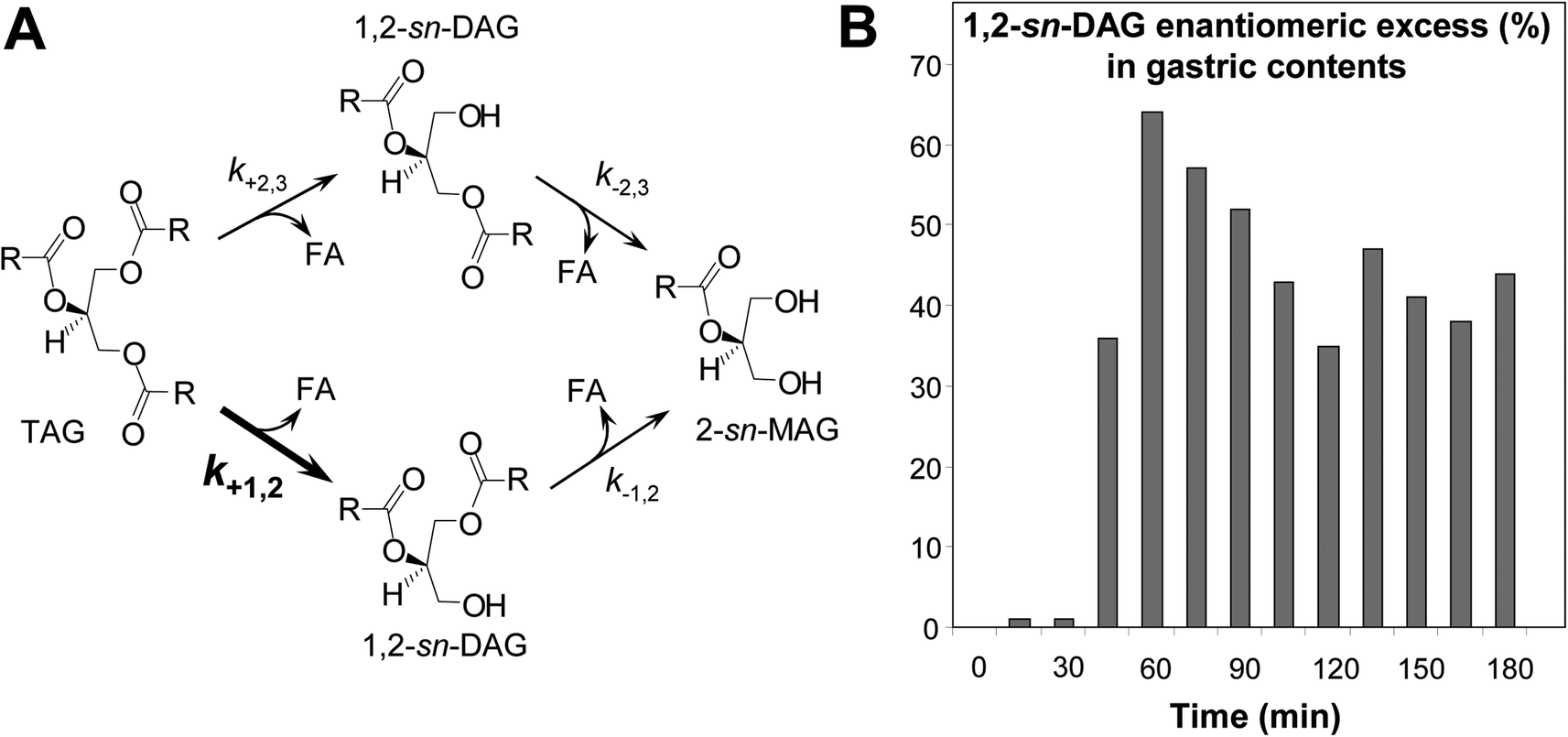 | ||
| Fig. 5 Stereoselective hydrolysis of TAG by gastric lipase. Panel A: reaction scheme for the enzymatic conversion of TAG into DAG and their subsequent conversion into MAG by lipases. The ksubscript symbols represent the specificity constants for the corresponding reactions. Panel B: DAG enantiomeric excess measured in gastric samples recovered from dog stomach in the course of test meal digestion. The liquid test meal contained only triolein as a prochiral triglyceride substrate. DAG enantiomeric excess (ee1,2%) was estimated using the following equation: ee1,2% = 100 × [1,2-sn-diolein − 2,3-sn-diolein]/[1,2-sn-diolein + 2,3-sn-diolein], taking into account the respective amounts of 1,2-sn-diolein (hydrolysis of sn-3 ester bond in triolein) and 2,3-sn-diolein (hydrolysis of sn-1 ester bond in triolein) in gastric samples, obtained after total lipid extraction, derivatization of DAG into diastereomeric carbamates and separation by HPLC. Adapted from ref. 32. | ||
The stereospecificity of gastric lipase is important for its biological function: HGL is the first lipase involved in the digestion of milk fat and its sn-3 stereopreference allows the release in the stomach of short and medium chain fatty acids that are specifically found at the sn-3 position of milk TAGs.98–100 This was shown for instance in gastric samples collected from premature infants who received mother milk.101 Although caprylic acid (C8:0) is found at low levels in human milk (0.07 ± 0.01% wt/wt of total milk fatty acids in this study;101Fig. 6A), the proportion of C8:0 in FFA found in gastric contents after 90 min of digestion was 50-fold higher (3.48 ± 1.53% wt/wt of total FFA; Fig. 6B) than the proportion of C8:0 in human milk fatty acids, indicating a preferential release of the fatty acids at the sn-3 position of milk TAGs by HGL. Fatty acids with chain length up to C12 can be absorbed in the stomach102,103 and the action of gastric lipase therefore allows an early uptake of short and medium chain fatty acids from milk. This could be a faster route to provide energy via fatty acids and supplementation of meals with medium chain triglycerides (MCT) is based on these findings. Nevertheless, the gastric absorption of medium chain fatty acids like C8:0 may be more important for the regulation of appetite through the acylation of the gastrointestinal hormone ghrelin. Ghrelin, produced by X-cells in the gastric corpus, initiates food uptake104 but also growth hormone release.110 Ghrelin needs to be acylated by C8:0 to be functional and this octanoylation is specifically ensured by ghrelin-O-acyltransferase (GOAT).105 C8:0 partly comes from the diet (milk, MCT) and is directly used for the acyl-modification of ghrelin.106,107 It is therefore assumed that gastric lipase plays an important role in the release of C8:0 for ghrelin acylation in the stomach. The mechanism by which C8:0 is absorbed and then used for ghrelin octanoylation has however not been studied in detail so far.
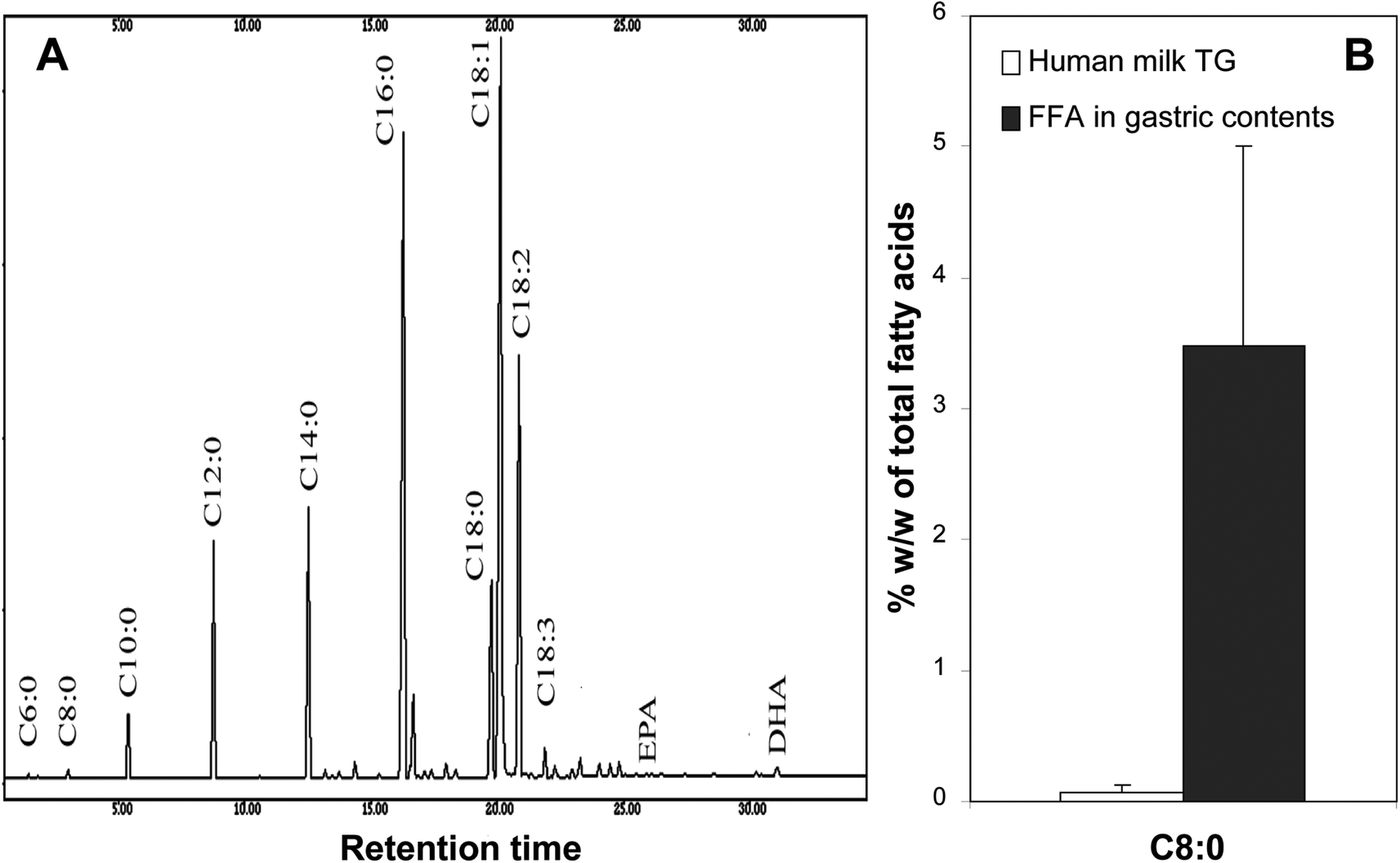 | ||
| Fig. 6 Selective release of C8:0 fatty acid from mother milk fat by gastric lipase in the stomach of newborns. Panel A: typical separation by gas chromatography of human milk fatty acids. Panel B: respective proportions of caprylic acid (C8:0) in human milk triglycerides (TAG) and free fatty acids recovered from the gastric content of newborns 90 minutes after meal ingestion. Adapted from ref. 106. | ||
A last and important property of gastric lipase is its resistance to pepsin. No degradation of HGL by pepsin is observed in gastric juice at pH values ranging from 2 to 7, i.e. the pH range in which HGL is highly stable and preserves its enzyme activity. Degradation of HGL by pepsin is only observed at pH 1 and below, and it occurs after HGL is first denatured by gastric acid.5 Therefore, the 3D structure of native HGL does not provide cleavage sites to pepsin. The removal of N-glycosylation sites in HGL (Asn15, Asn80, Asn252 and Asn308) by site-directed mutagenesis has revealed that N-glycosylation plays a role in the resistance to pepsin hydrolysis.108
Replacement of gastric lipase by other lipases
In summary, gastric lipase combines biochemical properties that make this enzyme rather unique among lipases: (i) stability and activity in the pH range 2 to 7 with an optimum around pH 5; (ii) high tensioactivity that allows resistance to bile salts and penetration into phospholipid layers covering TAG droplets; (iii) sn-3 stereospecificity for TAG hydrolysis; and (iv) resistance to pepsin. Most of these properties have been known for more than two decades and should provide a rational basis for the replacement of gastric lipase by other lipases when gastric lipase is not available.Among the microbial lipases that have been often used for in vitro gastric digestion, only the acid-resistant fungal lipase from Aspergillus niger has an optimum activity at acidic pH from 5 to 6.5, is active down to pH 2.5 and is resistant to pepsin (Table 4).109 It is however inhibited by bile salts,110 which indicates that it is a less tensioactive enzyme than gastric lipase, and its stereospecificity in TAG hydrolysis is unknown.
The sn-3 stereopreference of gastric lipase appears to be important for milk fat digestion in early life and probably for the digestion of dairy products like cheese in adults. Only a few lipases have been shown to display sn-3 stereospecificity. Using prochiral triolein as the substrate, several lipases have been ranked based on the enantiomeric excess of 1,2-sn-diolein vs. 2,3-sn-diolein (sn-3 stereopreference) or 2,3-sn-diolein vs. 1,2-sn-diolein (sn-1 stereopreference) measured at low TAG hydrolysis levels (3–6%; Table 3).29,111 Apart from gastric lipases, sn-3 stereopreference in TAG hydrolysis was reported for the plant lipase from Carica papaya latex (CPL),111 dog pancreatic lipase, fungal lipases from Fusarium solani and Penicillium simplicissimum, and bacterial lipases from Chromobacterium viscosum and Pseudomonas glumae.29 Only Fusarium solani cutinase was however found to display a high sn-3 stereospecificity like gastric lipases (Table 3). Among the sn-3 lipases, only CPL was tested under test meal conditions in vitro. CPL shows an optimum activity and stability at pH 6–7, in the presence and absence of bile, but its stability at pH below 5 is very low.112 It is resistant to many proteases like papain,113 but its resistance to pepsin has not been tested so far. Among the microbial lipases used for in vitro gastric digestion, lipases from Rhizopus oryzae (the same as Rhizopus arrhizus), Candida rugosa, Pseudomonas fluorescens and Rhizomucor miehei are all sn-1 stereospecific enzymes (Table 2)29,33,34 and therefore display a reversed stereospecificity compared to gastric lipase. The lipase from Aspergillus niger displays a 1,3-sn-regioselectivity in TAG hydrolysis but its stereospecificity towards sn-1 or sn-3 position was not demonstrated to our knowledge. One article reports sn-3 stereospecificity for a lipase from the Aspergillus niger NCIM 1207 strain but a careful reading reveals that sn-3 stereospecificity cannot be deduced from the thin layer chromatography analysis of triolein hydrolysis presented in this article.114 Indeed, the authors identified a band with the same retention time as 1,2-diolein used as the reference standard, and concluded that the ester bond at sn-3 position of triolein was cleaved by Aspergillus niger lipase, but 1,2-diolein and 2,3-diolein have the same retention time and cannot be distinguished using this separation method.114
| Lipasea | Regioselectivityb | Stereospecificityb | Optimum pHc | Inhibition by bile saltsd | Resistance to pepsin |
|---|---|---|---|---|---|
| n.d., not documenteda Some names of microbial species have changed and former names are indicated in parentheses.b Relative to TAG hydrolysis.c Relative to long chain TAG hydrolysis.d Since bile salts can be lipase activators at low concentration (<CMC) and lipase inhibitors at high concentration (>CMC),156,157 only the inhibition at bile salt concentration >CMC was considered here. | |||||
| Gastric lipase | No53 | sn-330 | 4–5.452,53,89 | No52,74 | Yes5 |
| Rhizopus oryzae (or arrhizus, or delemar) | 1,3158 | sn-129 | 7–8159 | Yes116,117 | Yes159 |
| Aspergillus niger | 1,3160 | n.d. | 5–6.5159,161 | Yes110 | Yes113,115 |
| Candida rugosa (or cylindracea) | No162 | sn-129 | 5–8161,163 | Yes,163 | No165 |
| No164 | |||||
| Pseudomonas fluorescens | 1,329 | sn-129 | 7–10166–170 | n.d. | n.d. |
| Rhizomucor miehei (or Mucor miehei) | 1,3171 | sn-129,33 | 7–8.2161 | n.d. | n.d. |
| Burkholderia cepacia (or Pseudomonas cepacia) | No171 | sn-1172–174 | 8175,176 | n.d. | n.d. |
| Lipases | Stereopreference | 1,2-ee (%) | 2,3-ee (%) |
|---|---|---|---|
| Dog gastric lipase | sn-3 | 76.2 | — |
| Human gastric lipase | sn-3 | 73.0 | — |
| Rabbit gastric lipase | sn-3 | 46.4 | — |
| Candida antarctica A lipase | sn-1 | — | 18.5 |
| Candida antarctica B lipase | sn-1 | — | 38.5 |
| Carica papaya lipase | sn-3 | 17.0 | — |
| Candida rugosa lipase | sn-1 | — | 18.5 |
| Chromobacterium viscosum lipase | sn-3 | 21.5 | — |
| Dog pancreatic lipase | sn-3 | 9.0 | — |
| Fusarium solani cutinase | sn-3 | 71.9 | — |
| Geotrichum candidum A lipase | sn-1 | — | 28.3 |
| Geotrichum candidum B lipase | sn-1 | — | 24.8 |
| Thermomyces lanuginosus lipase | sn-1 | — | 26.0 |
| Rhizomucor miehei lipase | sn-1 | — | 88.6 |
| Pseudomonas aeruginosa lipase | sn-1 | — | 15.8 |
| Penicillium camemberti lipase | sn-1 | — | 64.7 |
| Pseudomonas fluorescens lipase | sn-1 | — | 68.1 |
| Pseudomonas glumae lipase | sn-3 | 25.0 | — |
| Penicillium simplicissimum lipase | sn-3 | 30.5 | — |
| Rhizopus arrhizus (oryzae) lipase | sn-1 | — | 57.5 |
| Lipases | k +1,2 | k +2,3 | k −1,2 | k −2,3 |
|---|---|---|---|---|
| Dog gastric lipase | 3.55 | 1.00 | 3.70 | 1.39 |
| Dog pancreatic lipase | 1.25 | 1.00 | 1.01 | 0.75 |
| Porcine pancreatic lipase | 1.00 | 1.00 | 1.74 | 1.70 |
| Rhizomucor miehei lipase | 0.16 | 1.00 | 0.15 | 2.16 |
| Yarrowia lipolytica LIP2 lipase | 1.08 | 1.00 | 1.93 | 1.69 |
Other preduodenal lipases from the acid lipase gene family115,116 and various mammalian species might be used for replacing human gastric lipase. Depending upon the species investigated,117,118 preduodenal lipase is synthesized and secreted either by the von Ebner glands of the posterior area of the tongue (lingual lipase in small rodents) or the oro-pharyngeal tissues (pregastric or pharyngeal lipase in young ruminants) or in the gastric mucosa (gastric lipase in omnivores and monogastric herbivores) (Fig. 7 and Table 5). Whatever the species, preduodenal lipase is produced from a gene orthologous to the gene of HGL in humans (LIPF).77,115,116,119,120 The pregastric lipase of ruminants is found in young animals (calf, kid goat, lamb) during the lactation period and is responsible for the hydrolysis of milk fat globules in the abomasum (stomach).121–123 In addition to this physiological role, pregastric lipases of ruminants have been used in the dairy industry for various applications: enhancing the flavour of cheeses, accelerating cheese ripening, manufacturing cheese-like products and lipolysing butterfat and cream.123–125 Each type of pregastric lipase, depending upon the animal species, gives rise to its own characteristic flavour profile due to the fatty acid and/or regio- and stereoselectivity of the corresponding enzyme.126,127 Commercial pregastric lipases are available from various suppliers (DSM for Capalase® K and Capalase® KL lipases; CHR Hansen for Lipase Kid-Goat ST20, Lipase Calf 57 LFU, Spice IT™ AC and Spice IT™ AG; DuPont Danisco, Clerici-Sacco) in the form of liquid extracts, pastes and vacuum or freeze dried powders. Studies on pregastric lipases have often been carried out with these crude enzyme preparations.128 Only a few kinetic and biochemical studies have been performed with the enzymes purified from either the epiglottis/gullet, the GI tract region bordered anteriorly by the vallate papillae of the tongue and posteriorly by the pharyngeal end of the oesophagus129–133 or complete rennet paste.134 In the latter case, it is not always clear from the literature whether the enzyme characterized and named pregastric esterase is identical to the acid pregastric lipase. Like gastric lipases, the pregastric lipases of ruminants show an optimum activity at low pH (4 for lamb and 5–6 for calf) and their activity can be measured in the presence of bile salts.117,130 These enzymes may therefore be used for in vitro digestion studies and their abundance (Table 5) in animals present in the human food chain suggests the possibility to source these enzymes as by-products of meat production. One limitation of the use of pregastric lipases of ruminants might be however their resistance to acidic conditions. Indeed, these lipases are less resistant to acid denaturation than gastric lipase with a pH threshold for enzyme inactivation at around 3.5 while gastric and lingual lipases remain stable down to pH 1.5 in general.117 One article reports the use of calf pregastric lipase in an in vitro model of gastric digestion but the enzyme stability was found to be low and the reproducibility of results was impaired.135
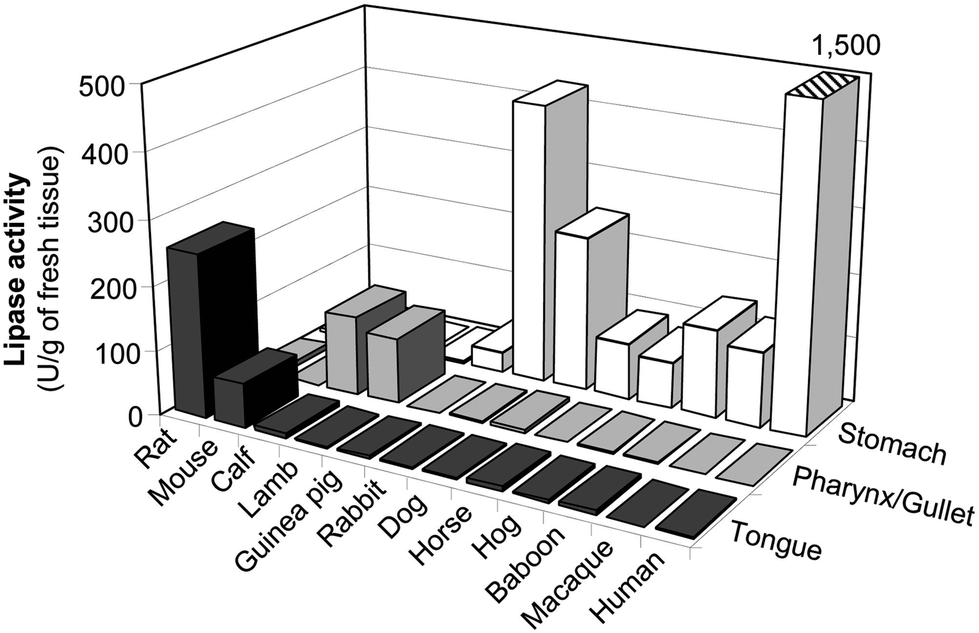 | ||
| Fig. 7 Tissular localization of preduodenal lipase activities in different mammalian species. Lipase activities were measured with the assay conditions optimized for gastric lipase and are expressed in units (U) per g of fresh tissue, with 1 U = 1 μmol of fatty acid released per minute. Adapted from ref. 72 and 117. | ||
| Species | Localization | Lipase activity | |
|---|---|---|---|
| U per organ | U per kg body weight | ||
| Rat | Tongue | 73 | 243 |
| Mouse | Tongue | 7 | 100 |
| Calf | Pharynx/gullet | 13130 |
73 |
| Lamb | Pharynx/gullet | 3000 | 55 |
| Guinea pig | Stomach | 432 | 864 |
| Rabbit | Stomach | 7130 | 3240 |
| Dog | Stomach | 13590 |
230 |
| Horse | Stomach | 30860 |
62 |
| Hog | Stomach | 5185 | 52 |
| Baboon | Stomach | 1315 | 88 |
| Macaque | Stomach | 2110 | 210 |
| Human | Stomach | 164000 |
2340 |
Production of gastric lipase
Gastric lipase has been obtained and characterized from various sources because it is a candidate for pancreatic enzyme replacement therapy (PERT) in patients with pancreatic exocrine insufficiency (PEI).76,136,137 Most advances in the biochemical characterization of gastric lipase have been obtained from the enzyme (HGL) purified from human gastric juice138 or stomachs from various species like rabbits89 and dogs.53 Although purified HGL has been tested in vitro under test meal conditions and compared to human gastric juice,38,63,139 the use of such an enzyme is limited by the availability of gastric juice and ethical considerations. The production of recombinant gastric lipase as an alternative and sustainable source of enzyme has been explored since the 1980s. An active recombinant human gastric lipase (rHGL) was found to be produced in the yeasts Saccharomyces cerevisiae77,140 and Schizosaccharomyces pombe,141,142 in Cos-7 and human embryonal kidney (HEK-293) cells,123,124 in insect cells,120,121 as well as Nicotiana benthamiana leaves using a transient expression system.143 Yeast and insect cell systems were not suitable however for industrial production since the lipase either remained stacked to the yeast cell wall76,120 or was produced at low levels (<40 mg L−1 of culture).140,144,145 Recombinant dog gastric lipase (rDGL) was also produced in insect cells using a baculovirus expression vector (35 mg L−1 of culture).146 Production using baculovirus-infected insect cells was however not continuous and led to a protein with lower glycosylation compared to native gastric lipase and poor resistance to pepsin.108 In addition to high production costs, the biochemical properties of rHGL produced in insect cells preclude its use for in vitro digestion in the presence of pepsin. rHGL produced in tobacco leaves appeared to be highly resistant to pepsin and reasonable production yields could be envisioned.143 Production yield in Cos-7 and HEK-293 cells has not been documented147,148 but producing a recombinant protein in these cells is usually more expensive than in yeast, fungi or bacteria.Whatever the application of recombinant gastric lipase in PERT or in vitro digestion, the enzyme should be produced at a low cost, similar to that of porcine pancreatic extracts. This was achieved with the production of a recombinant dog gastric lipase (rDGL) in transgenic plants, in which therapeutic proteins can be produced at reasonable cost (US $5.90–43 per g) compared to the same proteins produced in mammalian cells (US $300–3000 per g).149 DGL was selected because it was the lipase showing the highest lipolytic activity on long chain TAGs at low pH levels, with an optimum activity at pH 4.53 The cDNA encoding DGL150 was used to transform first tobacco151,152 and then corn.80,149 rDGL was produced at high levels in both tobacco leaves and corn seeds (approx. 1 g kg−1 of corn; personal communication from Dr Dominique Mison, Meristem Therapeutics SA, Clermont-Ferrand, France). The production and clinical development of rDGL were however seriously impaired by concerns about GMO and transgenic plants in France and Meristem Therapeutics SA stopped its activities in 2008. Although rDGL is no longer in production today, a few batches have been available for in vitro digestion studies. rDGL was thus used for testing the gastric lipolysis of lipid-based drug delivery systems,64,87,88,153,154 of citric acid esters of mono- and diglycerides (CITREM) and CITREM-containing infant formula/emulsions67 and various emulsions of flaxseed oil.68
Another method to obtain gastric lipase is by purification from animal sources since animal and human gastric lipases share similar biochemical properties.53,89 The production of native gastric lipase from animal stomachs was investigated by the pharmaceutical industry (Jouveinal Laboratories, Fresnes, France) and CNRS in the 1980s, in order to produce a drug product similar to porcine pancreatic extracts for the treatment of PEI.136 After a screening of preduodenal lipases in different mammals,117 the rabbit was identified as one of the species with the highest level of lipase in gastric tissues, in relation to its size (Fig. 7 and Table 5). Since rabbit is present in the human food chain, it could be used as an available source of gastric lipase as a by-product of meat production. Protocols to prepare rabbit gastric extract (RGE)136 and further purified rabbit gastric lipase (RGL)89 were implemented. The clinical development of RGE as a drug of animal origin was later stopped when prion diseases potentially transmissible from animals to humans were discovered but, nevertheless, the feasibility of RGE production was established at that time. There is today new interest in RGE for in vitro digestion experiments, particularly because it contains both gastric lipase and pepsin and can be used as a global source of gastric enzymes, like porcine pancreatic extracts for pancreatic enzymes. A combination of RGE and porcine pancreatic extracts was tested in a two-step static digestion model and the in vitro gastrointestinal lipolysis of test meal TAG was found to be similar to the lipolysis obtained with either human gastric and pancreatic juices or purified HGL and HPL.65 More recently, purified RGL and pepsin were used for testing the gastric lipolysis and proteolysis of different milk formulas under premature infant conditions.39
Conclusion
The development of physiologically relevant in vitro models of gastric digestion requires that the gastric phase of lipolysis and the role of gastric lipase are taken into account. Gastric lipolysis is not only significant and triggers further action of pancreatic enzymes, but gastric lipolysis and proteolysis processes are tightly linked. The data collected in this review should help researchers in designing in vitro gastric digestion models with relevant pH values and lipases. So far, none of the microbial lipases used to replace gastric lipase have displayed the biochemical properties of this unique enzyme and their use should not be recommended. If one has to select single gastric conditions for a static digestion model, the use of pH conditions prevailing in the stomach at 50% gastric emptying (pH 5–5.5) makes more sense than using low pH values corresponding to fasting conditions or to low amounts of food into the stomach. This will mean however a major adaptation of the current practice, often consisting of a first incubation at low pH (gastric phase; mostly pH 2) before raising the pH to 6–7 for the intestinal phase of digestion. Under these conditions, gastric lipolysis, if any, is certainly extremely low and its impact on overall digestion and intestinal events may not be seen. These drastic changes in pH conditions may also affect the stability and structure of food and emulsions, in a manner that does not exist in vivo, and thus may create artifacts. Reconsidering the gastric phase of in vitro digestion will certainly imply re-evaluating the data previously obtained with static intestinal digestion models.Acknowledgements
The EIPL laboratory (UMR7282 CNRS-Aix Marseille Université) is a member of the LISA Carnot Institute (Lipids for health and industry) supported by Agence Nationale de la Recherche (ANR-07-CARN-009-01). The authors have no conflict of interest to disclose.References
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carriere, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Menard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. Wickham, W. Weitschies and A. Brodkorb, Food Funct., 2014, 5, 1113–1124 CAS.
- E. S. Kostewicz, B. Abrahamsson, M. Brewster, J. Brouwers, J. Butler, S. Carlert, P. A. Dickinson, J. Dressman, R. Holm, S. Klein, J. Mann, M. McAllister, M. Minekus, U. Muenster, A. Mullertz, M. Verwei, M. Vertzoni, W. Weitschies and P. Augustijns, Eur. J. Pharm. Sci., 2014, 57, 342–366 CrossRef CAS PubMed.
- H. D. Williams, P. Sassene, K. Kleberg, J. C. Bakala-N'Goma, M. Calderone, V. Jannin, A. Igonin, A. Partheil, D. Marchaud, E. Jule, J. Vertommen, M. Maio, R. Blundell, H. Benameur, F. Carriere, A. Mullertz, C. J. Porter and C. W. Pouton, J. Pharm. Sci., 2012, 101, 3360–3380 CrossRef CAS PubMed.
- F. Carriere, V. Raphel, H. Moreau, A. Bernadac, M. A. Devaux, R. Grimaud, J. A. Barrowman, C. Benicourt, J. L. Junien and R. Laugier, et al. , Gastroenterology, 1992, 102, 1535–1545 CAS.
- E. Ville, F. Carriere, C. Renou and R. Laugier, Digestion, 2002, 65, 73–81 CAS.
- P. B. Pedersen, P. Vilmann, D. Bar-Shalom, A. Müllertz and B. S. Baldursdottir, Eur. J. Pharm. Biopharm., 2013, 85, 958–965 CrossRef CAS PubMed.
- F. Carrière, P. Grandval, P. C. Gregory, C. Renou, F. Henniges, S. Sander-Struckmeier and R. Laugier, JOP, 2005, 6, 206–215 Search PubMed.
- R. Verger, G. H. de Haas, L. Sarda and P. Desnuelle, Biochim. Biophys. Acta, 1969, 188, 272–282 CrossRef CAS.
- P. Desnuelle, H. Sjöström and O. Norén, Molecular and cellular basis of digestion, Elsevier, Amsterdam–New York–Oxford, 1986 Search PubMed.
- D. P. Muller and G. K. Ghale, Ann. Clin. Biochem., 1982, 19, 89–93 CrossRef CAS PubMed.
- D. G. Kelly, B. Sternby and E. P. DiMagno, Gastroenterology, 1991, 100, 189–195 CAS.
- F. Carrière, C. Renou, E. Ville, P. Grandval and R. Laugier, Digestion, 2001, 64, 46–53 Search PubMed.
- F. Carriere, P. Grandval, C. Renou, A. Palomba, F. Prieri, J. Giallo, F. Henniges, S. Sander-Struckmeier and R. Laugier, Clin. Gastroenterol. Hepatol., 2005, 3, 28–38 CrossRef CAS PubMed.
- J. C. Bakala N'Goma, S. Amara, K. Dridi, V. Jannin and F. Carriere, Ther. Delivery, 2012, 3, 105–124 CrossRef.
- L. Marciani, Neurogastroenterol. Motil., 2011, 23, 399–407 CrossRef CAS PubMed.
- M. J. Ferrua and R. P. Singh, J. Food Sci., 2010, 75, R151–R162 CrossRef CAS PubMed.
- L. Lundin, M. Golding and T. J. Wooster, Nutr.Diet., 2008, 65, S79–S85 CrossRef.
- D. J. McClements and Y. Li, Food Funct., 2010, 1, 32–59 CAS.
- C. Bourlieu, O. Ménard, K. Bouzerzour, G. Mandalari, A. Macierzanka, A. R. Mackie and D. Dupont, Crit. Rev. Food Sci. Nutr., 2014, 54, 1427–1457 CrossRef PubMed.
- E. Abrahamse, M. Minekus, G. A. van Aken, B. van de Heijning, J. Knol, N. Bartke, R. Oozeer, E. M. van der Beek and T. Ludwig, Food Dig., 2012, 3, 63–77 CrossRef CAS PubMed.
- S. J. Hur, B. O. Lim, E. A. Decker and D. J. McClements, Food Chem., 2011, 125, 1–12 CrossRef CAS.
- M. Smeets-Peeters, T. Watson, M. Minekus and R. Havenaar, Nutr. Res. Rev., 1998, 11, 45–69 CrossRef CAS PubMed.
- G. M. Bornhorst and R. Paul Singh, Annu. Rev. Food Sci. Technol., 2014, 5, 111–132 CrossRef CAS PubMed.
- T. R. Van de Wiele, A. G. Oomen, J. Wragg, M. Cave, M. Minekus, A. Hack, C. Cornelis, C. J. Rompelberg, L. L. De Zwart, B. Klinck, J. Van Wijnen, W. Verstraete and A. J. Sips, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1203–1211 CrossRef CAS PubMed.
- J. Chen, V. Gaikwad, M. Holmes, B. Murray, M. Povey, Y. Wang and Y. Zhang, Food Funct., 2011, 2, 174–182 CAS.
- M. J. S. Wickham, R. M. Faulks, J. Mann and G. Mandalari, Dissolution Technol., 2012, 19, 15–22 CrossRef CAS.
- F. Kong and R. P. Singh, J. Food Sci., 2010, 75, E627–E635 CrossRef CAS PubMed.
- F. Carriere, J. A. Barrowman, R. Verger and R. Laugier, Gastroenterology, 1993, 105, 876–888 CAS.
- E. Rogalska, C. Cudrey, F. Ferrato and R. Verger, Chirality, 1993, 5, 24–30 CrossRef CAS PubMed.
- E. Rogalska, S. Ransac and R. Verger, J. Biol. Chem., 1990, 265, 20271–20276 CAS.
- S. Ransac, E. Rogalska, Y. Gargouri, A. M. T. J. Deveer, F. Paltauf, G. H. de Haas and R. Verger, J. Biol. Chem., 1990, 265, 20263–20270 CAS.
- F. Carrière, E. Rogalska, C. Cudrey, F. Ferrato, R. Laugier and R. Verger, Bioorg. Med. Chem., 1997, 5, 429–435 CrossRef.
- J. A. Rodriguez, L. D. Mendoza, F. Pezzotti, N. Vanthuyne, J. Leclaire, R. Verger, G. Buono, F. Carriere and F. Fotiadu, Anal. Biochem., 2008, 375, 196–208 CrossRef CAS PubMed.
- D. A. Mitchell, J. A. Rodriguez, F. Carriere and N. Krieger, J. Biotechnol., 2008, 135, 168–173 CrossRef CAS PubMed.
- I. Douchet, G. De Haas and R. Verger, Chirality, 2003, 15, 220–226 CrossRef CAS PubMed.
- C. Bourlieu, F. Rousseau, V. Briard-Bion, M. N. Madec and S. Bouhallab, Food Res. Int., 2012, 49, 533–544 CrossRef CAS.
- C. Villemejane, R. Wahl, P. Aymard, S. Denis and C. Michon, Food Chem., 2015, 182, 55–63 CrossRef CAS PubMed.
- F. Carriere, C. Renou, V. Lopez, J. De Caro, F. Ferrato, H. Lengsfeld, A. De Caro, R. Laugier and R. Verger, Gastroenterology, 2000, 119, 949–960 CrossRef CAS.
- C. Bourlieu, O. Ménard, A. De La Chevasnerie, L. Sams, F. Rousseau, M.-N. Madec, B. Robert, A. Deglaire, S. Pezennec, S. Bouhallab, F. Carrière and D. Dupont, Food Chem., 2015, 182, 224–235 CrossRef CAS PubMed.
- M. J. Roman, B. J. Burri and R. P. Singh, J. Agric. Food Chem., 2012, 60, 9659–9666 CrossRef CAS PubMed.
- G. M. Bornhorst, M. J. Roman, K. C. Dreschler and R. P. Singh, Food Biophys., 2014, 9, 39–48 CrossRef.
- C. Lueamsaisuk, R. Lentle, A. MacGibbon, L. Matia-Merino and M. Golding, Food Hydrocolloids, 2015, 43, 785–793 CrossRef CAS.
- T. J. Wooster, L. Day, M. Xu, M. Golding, S. Oiseth, J. Keogh and P. Clifton, Food Hydrocolloids, 2014, 36, 102–114 CrossRef CAS.
- K. Larsson, L. Cavonius, M. Alminger and I. Undeland, J. Agric. Food Chem., 2012, 60, 7556–7564 CrossRef CAS PubMed.
- S. K. Marmon and I. Undeland, Food Chem., 2013, 138, 214–219 CrossRef CAS PubMed.
- V. L. Go, A. F. Hofmann and W. H. Summerskill, Gastroenterology, 1970, 58, 321–328 CAS.
- J. R. Malagelada, G. F. Longstreth, W. H. Summerskill and V. L. Go, Gastroenterology, 1976, 70, 203–210 CAS.
- C. Renou, F. Carrière, E. Ville, P. Grandval, M. Joubert-Collin and R. Laugier, Digestion, 2001, 63, 207–213 CAS.
- P. Langguth, K. M. Lee, H. Spahn-Langguth and G. L. Amidon, Biopharm. Drug Dispos., 1994, 15, 719–746 CrossRef CAS PubMed.
- S. Lartigue, Y. Bizais, S. B. Des Varannes, A. Murat, B. Pouliquen and J. P. Galmiche, Dig. Dis. Sci., 1994, 39, 109–115 CrossRef CAS PubMed.
- C. M. Brophy, J. G. Moore, P. E. Christian, M. J. Egger and A. T. Taylor, Dig. Dis. Sci., 1986, 31, 799–806 CrossRef CAS PubMed.
- Y. Gargouri, G. Pieroni, C. Riviere, J. F. Sauniere, P. A. Lowe, L. Sarda and R. Verger, Gastroenterology, 1986, 91, 919–925 CAS.
- F. Carriere, H. Moreau, V. Raphel, R. Laugier, C. Benicourt, J. L. Junien and R. Verger, Eur. J. Biochem., 1991, 202, 75–83 CrossRef CAS PubMed.
- F. Carrière, Y. Gargouri, H. Moreau, S. Ransac, E. Rogalska and R. Verger, in Lipases: Their structure, biochemistry and application, ed. P. Wooley and S. B. Petersen, Cambridge University Press, Cambridge, England, 1994, pp. 181–205 Search PubMed.
- J. B. Jansen, M. Fried, W. P. Hopman, C. B. Lamers and J. H. Meyer, Dig. Dis. Sci., 1994, 39, 571–576 CrossRef CAS PubMed.
- M. Fried, E. A. Mayer, S. R. Bloom, I. L. Taylor and J. H. Meyer, Regul. Pept., 1989, 26, 305–312 CrossRef CAS PubMed.
- M. Fried, E. A. Mayer, J. B. Jansen, C. B. Lamers, I. L. Taylor, S. R. Bloom and J. H. Meyer, Gastroenterology, 1988, 95, 1344–1350 CAS.
- S. Moberg and G. Carlberger, Scand. J. Gastroenterol., 1974, 9, 29–32 CAS.
- J. N. Hunt and D. F. Stubbs, J. Physiol., 1975, 245, 209–225 CrossRef CAS.
- J. N. Hunt, Am. J. Physiol., 1980, 239, G1–G4 CAS.
- J. G. Moore, P. E. Christian and R. E. Coleman, Dig. Dis. Sci., 1981, 26, 16–22 CrossRef CAS PubMed.
- J. N. Hunt, J. L. Smith and C. L. Jiang, Gastroenterology, 1985, 89, 1326–1330 CAS.
- F. Carriere, C. Renou, S. Ransac, V. Lopez, J. De Caro, F. Ferrato, A. De Caro, A. Fleury, P. Sanwald-Ducray, H. Lengsfeld, C. Beglinger, P. Hadvary, R. Verger and R. Laugier, Am. J. Physiol. Gastrointest. Liver Physiol., 2001, 281, G16–G28 CAS.
- S. Fernandez, S. Chevrier, N. Ritter, B. Mahler, F. Demarne, F. Carriere and V. Jannin, Pharm. Res., 2009, 26, 1901–1910 CrossRef CAS PubMed.
- P. Capolino, C. Guérin, J. Paume, J. Giallo, J.-M. Ballester, J.-F. Cavalier and F. Carrière, Food Dig., 2011, 2, 43–51 CrossRef CAS.
- C. Vors, P. Capolino, C. Guerin, E. Meugnier, S. Pesenti, M. A. Chauvin, J. Monteil, N. Peretti, M. Cansell, F. Carriere and M. C. Michalski, Food Funct., 2012, 3, 537–546 CAS.
- S. Amara, A. Patin, F. Giuffrida, T. J. Wooster, S. K. Thakkar, A. Benarouche, I. Poncin, S. Robert, V. Point, S. Molinari, H. Gaussier, S. Diomande, F. Destaillats, C. Cruz-Hernandez and F. Carriere, Food Funct., 2014, 5, 1409–1421 CAS.
- L. Couedelo, S. Amara, M. Lecomte, E. Meugnier, J. Monteil, L. Fonseca, G. Pineau, M. Cansell, F. Carriere, M. C. Michalski and C. Vaysse, Food Funct., 2015, 6, 1726–1735 CAS.
- Z. Szafran, H. Szafran, T. Popiela and G. Trompeter, Digestion, 1978, 18, 310–318 CAS.
- H. Moreau, J. F. Sauniere, Y. Gargouri, G. Pieroni, R. Verger and H. Sarles, Scand. J. Gastroenterol., 1988, 23, 1044–1048 CrossRef CAS PubMed.
- J. Moreau, M. Bouisson, D. Balas, A. Ravaud, S. Stupnik, L. Buscail, N. Vaysse and A. Ribet, Gastroenterology, 1990, 99, 175–180 CAS.
- H. Moreau, R. Laugier, Y. Gargouri, F. Ferrato and R. Verger, Gastroenterology, 1988, 95, 1221–1226 CAS.
- H. Moreau, A. Bernadac, Y. Gargouri, F. Benkouka, R. Laugier and R. Verger, Histochemistry, 1989, 91, 419–423 CAS.
- H. Lengsfeld, G. Beaumier-Gallon, H. Chahinian, A. De Caro, R. Verger, R. Laugier and F. Carrière, in Lipases and Phospholipases in Drug Development, ed. G. Müller and S. Petry, Wiley-VCH, Weinheim, 2004, pp. 195–229 Search PubMed.
- A. Bernadac, H. Moreau and R. Verger, Eur. J. Cell Biol., 1991, 55, 149–157 CAS.
- A. Aloulou and F. Carriere, Cell. Mol. Life Sci., 2008, 65, 851–854 CrossRef CAS PubMed.
- M. W. Bodmer, S. Angal, G. T. Yarranton, T. J. Harris, A. Lyons, D. J. King, G. Pieroni, C. Riviere, R. Verger and P. A. Lowe, Biochim. Biophys. Acta, 1987, 909, 237–244 CrossRef CAS.
- A. Roussel, S. Canaan, M. P. Egloff, M. Riviere, L. Dupuis, R. Verger and C. Cambillau, J. Biol. Chem., 1999, 274, 16995–17002 CrossRef CAS PubMed.
- H. Moreau, C. Abergel, F. Carrière, F. Ferrato, J. C. Fontecilla-Camps, C. Cambillau and R. Verger, J. Mol. Biol., 1992, 225, 147–153 CrossRef CAS PubMed.
- A. Roussel, N. Miled, L. Berti-Dupuis, M. Riviere, S. Spinelli, P. Berna, V. Gruber, R. Verger and C. Cambillau, J. Biol. Chem., 2002, 277, 2266–2274 CrossRef CAS PubMed.
- S. Canaan, A. Roussel, R. Verger and C. Cambillau, Biochim. Biophys. Acta, 1999, 1441, 197–204 CrossRef CAS.
- S. Vaganay, G. Joliff, O. Bertaux, E. Toselli, M. D. Devignes and C. Bénicourt, DNA Sequence, 1998, 8, 257–262 CAS.
- D. M. Blow, in The enzymes, ed. P. D. Boyer, Academic Press, New York and London, 1971, vol. III, pp. 185–212 Search PubMed.
- H. Chahinian, T. Snabe, C. Attias, P. Fojan, S. B. Petersen and F. Carrière, Biochemistry, 2006, 45, 993–1001 CrossRef CAS PubMed.
- A. Benarouche, V. Point, G. Parsiegla, F. Carriere and J. F. Cavalier, Colloids Surf., B, 2013, 111C, 306–312 CrossRef PubMed.
- Y. Pafumi, D. Lairon, P. Lechene de la Porte, C. Juhel, J. Storch, M. Hamosh and M. Armand, J. Biol. Chem., 2002, 277, 28070–28079 CrossRef CAS PubMed.
- S. Fernandez, V. Jannin, J. D. Rodier, N. Ritter, B. Mahler and F. Carriere, Biochim. Biophys. Acta, 2007, 1771, 633–640 CrossRef CAS PubMed.
- S. Fernandez, J. D. Rodier, N. Ritter, B. Mahler, F. Demarne, F. Carriere and V. Jannin, Biochim. Biophys. Acta, 2008, 1781, 367–375 CrossRef CAS PubMed.
- H. Moreau, Y. Gargouri, D. Lecat, J. L. Junien and R. Verger, Biochim. Biophys. Acta, 1988, 960, 286–293 CrossRef CAS.
- Y. Gargouri, G. Pieroni, P. A. Lowe, L. Sarda and R. Verger, Eur. J. Biochem., 1986, 156, 305–310 CrossRef CAS PubMed.
- S. Bezzine, F. Ferrato, M. G. Ivanova, V. Lopez, R. Verger and F. Carriere, Biochemistry, 1999, 38, 5499–5510 CrossRef CAS PubMed.
- H. van Tilbeurgh, M.-P. Egloff, C. Martinez, N. Rugani, R. Verger and C. Cambillau, Nature, 1993, 362, 814–820 CrossRef CAS PubMed.
- L. De la Fournière, M. G. Ivanova, J.-P. Blond, F. Carrière and R. Verger, Colloids Surf., B, 1994, 2, 585–593 CrossRef.
- Y. Gargouri, G. Pieroni, C. Riviere, P. A. Lowe, J. F. Sauniere, L. Sarda and R. Verger, Biochim. Biophys. Acta, 1986, 879, 419–423 CrossRef CAS.
- S. Bernback, L. Blackberg and O. Hernell, J. Clin. Invest., 1990, 85, 1221–1226 CrossRef CAS PubMed.
- S. Bernback, L. Blackberg and O. Hernell, Biochim. Biophys. Acta, 1989, 1001, 286–293 CrossRef CAS.
- P. Villeneuve, M. Pina, D. Montet and J. Graille, Chem. Phys. Lipids, 1995, 76, 109–113 CrossRef CAS.
- W. W. Christie and J. L. Clapperton, Int. J. Dairy Technol., 1982, 35, 22–24 CrossRef CAS.
- D. Otterby, H. Ramsey and G. Wise, J. Dairy Sci., 1964, 47, 993–996 CrossRef CAS.
- H. F. Helander and T. Olivecrona, Gastroenterology, 1970, 59, 22–35 CAS.
- C. Roman, F. Carriere, P. Villeneuve, M. Pina, V. Millet, U. Simeoni and J. Sarles, Pediatr. Res., 2007, 61, 83–88 CrossRef PubMed.
- J. P. Perret, J. Physiol. (Paris), 1980, 76, 159–166 CAS.
- J. P. Perret, J. Physiol. (Paris), 1982, 78, 221–230 CAS.
- D. E. Cummings and M. H. Shannon, Arch. Surg., 2003, 138, 389–396 CrossRef CAS PubMed.
- O. Al Massadi, M. H. Tschop and J. Tong, Peptides, 2011, 32, 2301–2308 CrossRef CAS PubMed.
- Y. Nishi, H. Hiejima, H. Hosoda, H. Kaiya, K. Mori, Y. Fukue, T. Yanase, H. Nawata, K. Kangawa and M. Kojima, Endocrinology, 2005, 146, 2255–2264 CrossRef CAS PubMed.
- Y. Nishi, H. Mifune and M. Kojima, Methods Enzymol., 2012, 514, 303–315 CAS.
- C. Wicker-Planquart, S. Canaan, M. Riviere and L. Dupuis, Eur. J. Biochem., 1999, 262, 644–651 CrossRef CAS PubMed.
- P. L. Zentler-Munro, B. A. Assoufi, K. Balasubramanian, S. Cornell, D. Benoliel, T. C. Northfield and M. E. Hodson, Pancreas, 1992, 7, 311–319 CrossRef CAS PubMed.
- H. Kermanshahi, D. D. Maenz and H. L. Classen, Poult. Sci., 1998, 77, 1671–1677 CrossRef CAS PubMed.
- E. Cambon, J. A. Rodriguez, M. Pina, V. Arondel, F. Carriere, F. Turon, J. Ruales and P. Villeneuve, Biotechnol. Lett., 2008, 30, 769–774 CrossRef CAS PubMed.
- S. Abdelkafi, B. Fouquet, N. Barouh, S. Durner, M. Pina, F. Scheirlinckx, P. Villeneuve and F. Carrière, Food Chem., 2009, 115, 488–494 CrossRef CAS.
- S. Abdelkafi, N. Barouh, B. Fouquet, I. Fendri, M. Pina, F. Scheirlinckx, P. Villeneuve and F. Carriere, Plant Foods Hum. Nutr., 2011, 66, 34–40 CrossRef CAS PubMed.
- N. C. Mhetras, K. B. Bastawde and D. V. Gokhale, Bioresour. Technol., 2009, 100, 1486–1490 CrossRef CAS PubMed.
- P. Lohse, S. Chahrokh-Zadeh and D. Seidel, J. Lipid Res., 1997, 38, 880–891 CAS.
- R. S. Holmes, L. A. Cox and J. L. VandeBerg, Comp. Biochem. Physiol., Part D: Genomics Proteomics, 2010, 5, 217–226 CrossRef PubMed.
- H. Moreau, Y. Gargouri, D. Lecat, J. L. Junien and R. Verger, Biochim. Biophys. Acta, 1988, 959, 247–252 CrossRef CAS.
- S. J. de Nigris, M. Hamosh, D. K. Kasbedar, T. C. Lee and P. Hamosh, Biochim. Biophys. Acta, 1988, 958, 38–45 CrossRef.
- M. Y. J. Timmermans, H. Teuchy and L. P. M. Kupers, Gene, 1994, 147, 259–262 CrossRef CAS PubMed.
- A. J. P. Docherty, M. W. Bodmer, S. Angal, R. Verger, C. Rivière, P. A. Lowe, A. Lyons, J. S. Emtage and T. J. R. Harris, Nucleic Acids Res., 1985, 13, 1891–1903 CrossRef CAS PubMed.
- H. A. Ramsey and J. W. Young, J. Dairy Sci., 1961, 44, 2227–2231 CrossRef.
- D. E. Otterby, H. A. Ramsey and G. H. Wise, J. Dairy Sci., 1964, 47, 993–996 CrossRef CAS.
- J. H. Nelson, R. G. Jensen and R. E. Pitas, J. Dairy Sci., 1976, 60, 327–362 CrossRef.
- A. M. Bech, Bull. Int. Dairy Fed., 1992, 269, 24–28 CAS.
- P. Birschbach, Bull. Int. Dairy Fed., 1992, 269, 36–39 CAS.
- P. Villeneuve, M. Pina and J. Graille, Chem. Phys. Lipids, 1996, 83, 161–168 CrossRef CAS PubMed.
- J. K. Ha and R. C. Lindsay, J. Dairy Sci., 1993, 76, 677–690 CrossRef CAS PubMed.
- H. A. Ramsey, G. H. Wise and S. B. Tove, J. Dairy Sci., 1956, 39, 1312–1322 CrossRef CAS.
- S. Bernback, O. Hernell and L. Blackberg, Eur. J. Biochem., 1985, 148, 233–238 CrossRef CAS PubMed.
- J. De Caro, F. Ferrato, R. Verger and A. de Caro, Biochim. Biophys. Acta, 1995, 1252, 321–329 CrossRef CAS.
- S. Bernback, O. Hernell and L. Blackberg, Biochim. Biophys. Acta, 1987, 922, 206–213 CrossRef CAS.
- B. J. Sweet, L. C. Matthews and T. Richardson, Arch. Biochem. Biophys., 1984, 234, 144–150 CrossRef CAS PubMed.
- C. J. O'Connor and R. D. Manuel, Prog. Colloid Polym. Sci., 1997, 106, 188–191 CrossRef.
- M. V. Calvo and J. Fontecha, J. Dairy Sci., 2004, 87, 1132–1142 CrossRef CAS PubMed.
- C. Lueamsaisuk, R. G. Lentle, A. K. H. MacGibbon, L. Matia-Merino and M. Golding, Food Hydrocolloids, 2014, 36, 162–172 CrossRef CAS.
- H. Moreau, R. Verger, D. Lecat and J. L. Junien. European Patent, EP0261016 A1, 1987 Search PubMed.
- C. Bénicourt, C. Blanchard, F. Carrière, R. Verger and J. L. Junien, in Clinical Ecology of Cystic Fibrosis, ed. H. Escobar, C. F. Baquero and L. Suárez, Elsevier Science Publishers, Amsterdam, 1993, pp. 291–295 Search PubMed.
- C. Tiruppathi and K. A. Balasubramanian, Biochim. Biophys. Acta, 1982, 712, 692–697 CrossRef CAS.
- P. Borel, M. Armand, P. Ythier, G. Dutot, C. Melin, M. Senft, H. Lafont and D. Lairon, J. Nutr. Biochem., 1994, 5, 124–133 CrossRef CAS.
- T. Crabbe, A. N. Weir, E. F. Walton, M. E. Brown, C. W. Sutton, N. Tretout, J. Bonnerjea, P. A. Lowe and G. T. Yarranton, Protein Expression Purif., 1996, 7, 229–236 CrossRef CAS PubMed.
- G. R. Smerdon, S. J. Aves and E. F. Walton, Gene, 1995, 165, 313–318 CrossRef CAS PubMed.
- G. R. Smerdon, E. F. Walton and S. J. Aves, Appl. Microbiol. Biotechnol., 1998, 49, 45–50 CrossRef CAS PubMed.
- M. Vardakou, F. Sainsbury, N. Rigby, F. Mulholland and G. P. Lomonossoff, Protein Expression Purif., 2012, 81, 69–74 CrossRef CAS PubMed.
- C. Wicker-Planquart, S. Canaan, M. Riviere, L. Dupuis and R. Verger, Protein Eng., 1996, 9, 1225–1232 CrossRef CAS PubMed.
- S. Canaan, L. Dupuis, M. Riviere, K. Faessel, J. L. Romette, R. Verger and C. Wicker-Planquart, Protein Expression Purif., 1998, 14, 23–30 CrossRef CAS PubMed.
- G. Joliff, S. Vaganay, C. Legay and C. Bénicourt, Biotechnol. Lett., 1998, 20, 697–702 CrossRef CAS.
- P. Lohse, S. Chahrokh-Zadeh, P. Lohse and D. Seidel, J. Lipid Res., 1997, 38, 892–903 CAS.
- P. Lohse, S. Chahrokh-Zadeh and D. Seidel, J. Lipid Res., 1997, 38, 1896–1905 CAS.
- Q. Zhong, Z. Gu and C. E. Glatz, J. Agric. Food Chem., 2006, 54, 8086–8092 CrossRef CAS PubMed.
- S. Vaganay, G. Joliff, O. Bertaux, E. Toselli, M. D. Devignes and C. Benicourt, DNA Sequence, 1998, 8, 257–262 CAS.
- V. Gruber, P. P. Berna, T. Arnaud, P. Bournat, C. Clément, D. Mison, B. Olagnier, L. Philippe, M. Theisen and S. Baudino, Mol. Breed., 2001, 7, 329–340 CrossRef CAS.
- N. Mokrzycki-Issartel, B. Bouchon, S. Farrer, P. Berland, H. Laparra, J. C. Madelmont and M. Theisen, FEBS Lett., 2003, 552, 170–176 CrossRef CAS PubMed.
- S. Fernandez, V. Jannin, S. Chevrier, Y. Chavant, F. Demarne and F. Carriere, Pharm. Res., 2013, 30, 3077–3087 CrossRef CAS PubMed.
- J. C. Bakala-N'Goma, H. D. Williams, P. J. Sassene, K. Kleberg, M. Calderone, V. Jannin, A. Igonin, A. Partheil, D. Marchaud, E. Jule, J. Vertommen, M. Maio, R. Blundell, H. Benameur, A. Mullertz, C. W. Pouton, C. J. Porter and F. Carriere, Pharm. Res., 2015, 32, 1279–1287 CrossRef PubMed.
- K. Thirstrup, F. Carriere, S. Hjorth, P. B. Rasmussen, H. Woldike, P. F. Nielsen and L. Thim, FEBS Lett., 1993, 327, 79–84 CrossRef CAS PubMed.
- V. Delorme, R. Dhouib, S. Canaan, F. Fotiadu, F. Carriere and J. F. Cavalier, Pharm. Res., 2011, 28, 1831–1842 CrossRef CAS PubMed.
- A. Aloulou, D. Puccinelli, A. De Caro, Y. Leblond and F. Carriere, Biochim. Biophys. Acta, 2007, 1771, 1446–1456 CrossRef CAS PubMed.
- M. Semeriva, G. Benzonana and P. Desnuelle, Bull. Soc. Chim. Biol., 1967, 49, 71–79 CAS.
- H. Kermanshahi, D. D. Maenz and H. L. Classen, Poult. Sci., 1998, 77, 1665–1670 CrossRef CAS PubMed.
- S. Okumura, M. Mieko Iwai and Y. Tsujisaka, Agric. Biol. Chem., 1976, 40, 655–660 CrossRef CAS.
- H. Uhlig, Industrial Enzymes and Their Applications, John Wiley & Sons, 1998 Search PubMed.
- G. Benzonana and S. Esposito, Biochim. Biophys. Acta, 1971, 231, 15–22 CrossRef CAS.
- N. Tomizuka, Y. Ota and K. Yamada, Agric. Biol. Chem., 1966, 30, 576–584 CrossRef CAS.
- H. Mtibaa, A. Fendri, A. Sayari, A. Ben Salah, H. Mejdoub and Y. Gargouri, Ol., Corps Gras, Lipides, 2002, 9, 49–53 CAS.
- S. Turki, G. Mrabet, Z. Jabloun, J. Destain, P. Thonart and H. Kallel, Biotechnol. Appl. Biochem., 2010, 57, 139–149 CrossRef CAS PubMed.
- R. Boran and A. Ugur, Prep. Biochem. Biotechnol., 2010, 40, 229–241 CrossRef CAS PubMed.
- K. Chakraborty and R. Paulraj, J. Agric. Food Chem., 2009, 57, 3859–3866 CrossRef CAS PubMed.
- P. F. Fox and L. Stepaniak, J. Dairy Res., 1983, 50, 77–89 CrossRef CAS PubMed.
- Y. P. Lee, G. H. Chung and J. S. Rhee, Biochim. Biophys. Acta, 1993, 1169, 156–164 CAS.
- Y. Kojima, M. Yokoe and T. Mase, Biosci., Biotechnol., Biochem., 1994, 58, 1564–1568 CrossRef CAS PubMed.
- R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608–1633 CrossRef.
- K.-E. Jaeger, S. Ransac, B. W. Dijkstra, C. Colson, M. Vanheuvel and O. Misset, FEMS Microbiol. Rev., 1994, 15, 29–63 CrossRef CAS PubMed.
- D. Lang and B. W. Dijkstra, Chem. Phys. Lipids, 1998, 93, 115–122 CrossRef CAS.
- D. A. Lang, M. L. Mannesse, G. H. de Haas, H. M. Verheij and B. W. Dijkstra, Eur. J. Biochem., 1998, 254, 333–340 CAS.
- G. da Silva Padilha, J. C. José Carlos Curvelo Santana, R. Monte Alegre and E. B. Tambourgi, Braz. Arch. Biol. Technol., 2012, 55, 7–19 CrossRef.
- M. Kordel, B. Hofmann, D. Schomburg and R. D. Schmid, J. Bacteriol., 1991, 173, 4836–4841 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fo00930h |
| This journal is © The Royal Society of Chemistry 2016 |