
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs ascorbate Dr Jekyll or Mr Hyde in the Cu(Aβ) mediated oxidative stress linked to Alzheimer's disease?†
Clémence
Cheignon
abc,
Fabrice
Collin
abc,
Peter
Faller‡
*ab and
Christelle
Hureau
*ab
aLCC (Laboratoire de Chimie de Coordination), CNRS UPR 8241, 205 route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: christelle.hureau@lcc-toulouse.fr
bUniversité de Toulouse; UPS, INPT, 31077 Toulouse, France
cUMR 152 Pharma Dev, Université de Toulouse, IRD, UPS, France
First published on 24th May 2016
Abstract
Evaluation of the pro versus antioxidant activity of ascorbate regarding Cu(Aβ) induced reactive oxygen species production in the context of Alzheimer's disease shows that a protective activity can only be observed at high ascorbate concentration for exogenous molecules but not for the amyloid-β peptide itself.
The misregulation of reactive oxygen species (ROS) leading to oxidative stress is observed in multiple diseases, including neurodegenerative disorders like Alzheimer's or Parkinson's disease. Therefore, there is strong interest in the effect of antioxidants as they could have potential therapeutic benefits.1 There are different families of antioxidants depending on their mode of action (sacrificial or catalytic scavengers of ROS produced, inhibitors of ROS production). Sacrificial scavengers work by capturing ROS and other free radical species via reduction or addition reactions.2 Ascorbate belongs to this family. It is a main physiological reducing agent present at a high concentration, in particular in the brain (up to 10 mM in neurons).3 It can reduce radicals and convert them into non-radicals, like for instance HO˙ to HO−, or O2˙− to H2O2. This is part of its antioxidant activity but transferring an electron to a non-radical species can also produce radicals. This happens when an electron is transferred to O2 or H2O2, leading to O2˙− or to the very reactive HO˙ radical. However, these reactions are not efficient,4 unless they are catalyzed. This can be performed by redox active metal ions like Cu(I/II) and Fe(II/III) which can catalyze very efficiently the transfer of a single electron. This is for instance the case in the well-known Fenton reaction leading to the formation of HO˙ from dioxygen and a reducing agent like ascorbate. Actually, redox active metal ions, mainly Fe(II/III), Cu(I/II) and Mn(II/III) play a crucial role in vivo that essentially depends on their coordination chemistry. This is illustrated by the role of Cu ions that can be catalytic scavengers of O2˙− when bound inside superoxide dismutase (SOD) or are involved in the catalytic production of ROS when loosely bound (Scheme 1).4
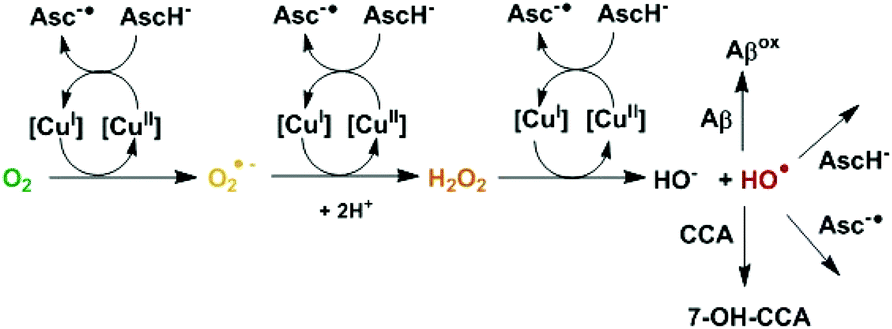 | ||
| Scheme 1 Mechanism of HO˙ production from ascorbate and dioxygen catalyzed by copper. HO˙ can then react with different compounds, with CCA to form the fluorescent 7-OH-CCA, or with ascorbate/ascorbyl or with Aβ in the case of Cu bound to Aβ. [Cu] corresponds to free or Aβ-bound Cu. | ||
In the case of the reaction of loosely bound Cu ions with ascorbate and dioxygen, it is known that the concentration of ascorbate has an important impact on the production of HO˙. In general, it was concluded that low concentrations of ascorbate are required for prooxidant conditions, while high concentrations are needed for antioxidant conditions.5 Thus there is a “cross-over” point, i.e. an ascorbate concentration at which the prooxidant ability of ascorbate is maximal. Such a value depends on the experimental conditions.5 This has been supported by experiments in different systems in vitro and in cellulo.6In vivo, the prooxidant activity of ascorbate seems to be limited to conditions where Fe and Cu are aberrantly coordinated and hence able to catalyze the Fenton type reaction.7
In Alzheimer's disease (AD), Cu ions are found in high concentrations (0.4 mM) in amyloid plaques, a hallmark of the disease.8 Cu is bound to the amyloid-β (Aβ) peptide in the extracellular plaques themselves but its coordination to the monomeric soluble form of Aβ is also proposed,9 as the Cu concentration can reach up to 10 μM in certain synaptic clefts.10,11 The coordination sites of Cu(II) and Cu(I) lie in the first 16 amino acids of the 40 to 42 amino-acid residue peptide. They have been extensively studied and are well characterized (Fig. S1 and S2†), in relation to their redox ability.9In vitro, Cu(Aβ) is able to catalyze the production of ROS in the presence of ascorbate, and it was proposed that these ROS can contribute to the oxidative stress observed in AD.12,13
In the present work, we have investigated the impact of Cu coordination to the Aβ peptide on modulation of the pro versus antioxidant properties of ascorbate. This is of interest with regard to therapeutic approaches in AD based on oxidative stress regulation. Coumarin-3-carboxylate (CCA) was used as a prototypical target mimicking the biological ones, to detect the deleterious radical HO˙. CCA reacts with HO˙ and forms the fluorescent adduct 7-hydroxycoumarin-3-carboxylate (7-OH-CCA), which shows emission at 450 nm upon excitation at 390 nm. While the detection of HO˙ by CCA was reported to be sensitive and specific,14 there are some parameters to be considered in order to have a quantitative detection (at a particular pH, see the ESI for details: Fig. S3–S6†).
In Fig. 1, black lines show the time dependence of Cu-induced formation of 7-OH-CCA in line with similar literature data.15–18 Two main phases were observed: (i) a first fast process with an exponential growth profile and (ii) a second slow process with an almost linear increase. The origin of the latter one is not known, but it seems to be independent of the Cu-catalyzed HO˙ production from ascorbate and O2 and thus irrelevant for the present study. Indeed, completion of the first process almost coincides with total ascorbate consumption (Fig. S7†) and thus there is not much ascorbate available when the second process occurs. Note that unless specified all the experiments described in the paper were performed with the C-terminally truncated peptide encompassing the first 16 amino-acid residues that correctly reproduce the binding properties of the full-length peptide.9
 | ||
| Fig. 1 Time dependence of HO˙ production measured by 7-OH-CCA fluorescence assay (λexcitation = 390 nm and λemission = 450 nm) for Cu (black curves), Aβ16 (dotted curves) and Cu-Aβ16 (grey curves) with ascorbate ((a) 0.5 mM, (b) 1 mM and (c) 5 mM). Phosphate-buffered solution (50 mM, pH 7.4) of CCA (0.5 mM), Cu (50 μM), either Aβ16 (60 μM) or no peptide. Experiments with only ascorbate give similar curves to Aβ16 with ascorbate (dotted curves). Arrows indicate the completion of ascorbate consumption as measured independently (Fig. S7†). | ||
In the presence of 1.2 equivalents of Aβ16, the curves are different (Fig. 1, grey curves). The first phase takes more time, indicating that the formation of 7-OH-CCA is slower, in line with the lower catalytic activity of Cu(Aβ16) with respect to ROS formation, described already in the past.15–17 Also the plateau reached during the second phase is lower compared to the one obtained in the presence of Cu only. This is in line with the observation that HO˙ reacts with the peptide Aβ itself thus precluding its reaction with CCA as previously observed.18–21 Indeed, the plateau corresponds to the part of HO˙ able to react with CCA, i.e. it reflects the part of HO˙ that would be able to damage biomolecules other than Aβ or ascorbate, like proteins, lipids or nucleic acids in a biological environment. In line with this comment, it is worth noting that the higher the ascorbate over the Aβ16 ratio, the closer the plateau obtained for free Cu and Cu(Aβ) species. At a high concentration of ascorbate, the first phase is more complex in the presence of a peptide with a sigmoid-like trend in line with (i) the oxidation of the Aβ16 peptide and (ii) the subsequent release of free Cu (inset in Fig. 1). This was verified by measuring the level of intact peptide by LC-MS (Fig. S8 and Table S1†).§ Indeed the peptide was degraded at the end of the reaction. The degradation degree depends on the ascorbate concentration, but is complete above 1 mM (Fig. S9†).
In order to determine the pro versus antioxidant regime of ascorbate with both free Cu and Cu(Aβ16), the ascorbate dependence of the 7-OH-CCA formation was measured and compared. Fig. 2¶ shows the initial rate (calculated over the first five minutes) of the 7-OH-CCA formation at different ascorbate concentrations for Cu and Cu(Aβ16). The initial rate can be considered as the most relevant reaction under steady state conditions of ascorbate and oxygen supposed to occur in vivo.
 | ||
| Fig. 2 Initial kinetics of 7-OH-CCA fluorescence at 450 nm, reflecting the scavenging of HO˙ by CCA. Phosphate-buffered solution (50 mM, pH 7.4) of CCA (0.5 mM) with Cu (left panel, (a) 10 μM, (b) 50 μM, (c) 100 μM), or Cu-Aβ16 (right panel, (a) 10–12 μM, (b) 50–60 μM, (c) 100–120 μM) and ascorbate (concentration between 0 and 5 mM). | ||
 | ||
| Fig. 3 Metal-catalyzed oxidation of CCA by HO˙. Phosphate-buffered solution (50 mM, pH 7.4) of CCA (0.5 mM) with Cu (left panel, (a) 10 μM, (b) 50 μM, (c) 100 μM), or Cu-Aβ16 (right panel, (a) 10–12 μM, (b) 50–60 μM, (c) 100–120 μM) and ascorbate (concentration between 0 and 5 mM). At the end of the ascorbate oxidation, POPSO buffer (pH 9.0, 0.4 M) is added to adjust the pH at 8.5. The fluorescence of 7-OH-CCA (λexcitation = 390 nm and λemission = 450 nm) is then measured. | ||
For free Cu, the shapes are similar for the three different Cu concentrations used and all show a very marked ascorbate concentration dependence. At low ascorbate concentration the initial rate of 7-OH-CCA formation increases with the ascorbate concentration. The maximal rate, which has previously been called the cross-over point,5 is observed around 0.7 mM in ascorbate. At higher ascorbate concentrations the initial rate of 7-OH-CCA formation decreases continuously, supporting an antioxidant property from 0.7 mM. This dependence can be explained due to the double effect of ascorbate: on one side, it promotes the production of HO˙ that further reacts with CCA (pro-oxidant part, Scheme 1) and on the other hand it scavenges HO˙ (antioxidant part).5 In the absence of ascorbate, no HO˙ is produced, as ascorbate is needed as an electron source to reduce dioxygen to HO˙. At low concentration, HO˙ trapped by CCA increases with ascorbate concentrations, because ascorbate fuels the formation of HO˙, and does not scavenge HO˙ efficiently. At higher ascorbate concentrations, the initial rate does not increase anymore, because the ascorbate is no more the limiting species and the scavenging ability becomes more and more efficient. For these reasons, the initial rate of 7-OH-CCA formation decreases.
In the presence of Aβ16, i.e. when HO˙ production is catalyzed by Cu(Aβ16), the overall dependence is similar, with a cross-over point at 0.7 mM of ascorbate (Fig. 2, right panel). In general, the initial Cu(Aβ16) induced formation of 7-OH-CCA formation is slower than without a peptide, in line with the more sluggish Cu(I/II) redox process of Cu when bound to Aβ.22–24 Note that only the very first points of the 7-OH-CCA formation curves were taken into account to avoid any issue due to the release of Cu at high ascorbate concentrations (inset in Fig. 1). The rate of 7-OH-CCA formation is dependent on the Cu(Aβ16) concentration. This is in line with the slower rate of HO˙ formation in the presence of Aβ16. The same trends were observed for the longer peptide encompassing the first 28 amino-acid residues and the full-length peptide (Fig. S10†).
Finally, the total amounts of 7-OH-CCA produced were measured. For doing this, the reaction was stopped after the total consumption of ascorbate (arrows in Fig. 1), and the intensities of the plateau were compared (Fig. 3) as a function of the initial ascorbate concentration and the Cu (resp. Cu(Aβ)) concentration.
For free Cu, the ascorbate concentration dependence follows a sort of saturation curve. Again this can be explained by the two properties of ascorbate that fuels the HO˙ production and scavenges them. At low concentration the formation of 7-OH-CCA increases steadily, as under this condition the former property prevails. At increasing concentrations the scavenging ability progressively counter-balances the formation of HO˙ and the highest ascorbate concentrations have no more effect on 7-OH-CCA formation. Note that a plateau is reached (and not the same shape curve as previously) because cumulative amounts of 7-OH-CCA (and not amounts of 7-OH-CCA at a given time) are measured in this case.
In the presence of Aβ a similar shape is obtained, but with much lower intensities. This is assigned to the scavenging ability of Aβ itself, which increases with the Cu(Aβ) concentration.
To probe the ability of ascorbate to protect the Aβ peptide, we have compared the degradation of the peptide after the same amount of HO˙ was produced, but at different initial ascorbate concentrations, i.e. (i) at the end of the reaction with 1 mM ascorbate and (ii) after consumption of 1 mM ascorbate with the initial concentration of ascorbate at 10 mM (Fig. S11†). This should yield the same amount of HO˙ produced, but in the second sample much more ascorbate was present to exert the antioxidant effect. Even under this condition, no rescue in Aβ degradation was observed (Table S2†). This indicated that although ascorbate can scavenge HO˙ before it reacts with CCA, it is much less efficient in intercepting HO˙ when reacting with the Aβ peptide itself. This could be explained by the fact that Aβ is linked to Cu ions and hence HO˙ are produced in the vicinity of the peptide. In contrast, CCA is not bound to the metal centre and has to encounter HO˙ by diffusion.
In the present communication, we have reported that ascorbate can have a pro- or an antioxidant effect concerning the initial rates of HO˙ attack on biological targets, here modelled by the CCA compound. The approach proposed here in the AD context with Cu(Aβ) species as the ROS production catalyst, could apply to any other chemical system able to form ROS. This includes other Cu-amyloidogenic peptide species, as encountered in Parkinson's disease, prion diseases, etc.25 In the present case, the “cross-over” point, i.e. the ascorbate concentration at which the rate of HO˙ formation is maximal, was observed at about 0.7 mM for a range of 10–100 μM Cu(Aβ). This means that below this concentration, ascorbate fuels the formation of HO˙ that mainly reacts with CCA and the Aβ peptide. At higher concentrations, HO˙ scavenging by ascorbate becomes more efficient and hence the rate of CCA oxidation slows down. Measuring the initial rate might be the most relevant condition in a more biological environment, as systems tend to maintain a steady state in terms of ascorbate and dioxygen concentrations. Under such conditions, the “cross-over” point corresponds to the maximal rate of HO˙ escaping from Cu(Aβ), and hence to maximal damage to biomolecules (proteins, DNA, lipids etc.) other than Aβ. At lower or higher ascorbate concentrations, less biomolecule damage would occur but for different reasons (weaker ROS production or higher scavenging activity, respectively). The situation for the Aβ peptide is different. Aβ scavenges the HO˙ due to its proximity with the place of ROS production and is oxidized regardless of ascorbate concentrations. Even very high ascorbate concentrations were not able to protect Aβ from oxidation, which is likely due to the proximity of the peptide residues to the HO˙ production site thus making it difficult for ascorbate to intercept the HO˙. However, our in vitro studies could mainly be relevant for the surroundings of Cu(Aβ) complexes and the general situation in vivo under AD conditions could be more complex and the local prooxidant activity of ascorbate could be counteracted.7
Sacrificial antioxidants have been investigated as therapeutic compounds in several neurodegenerative diseases including AD.19,26 In contrast to ascorbate, such compounds are mainly HO˙ scavengers but can hardly induce the formation of ROS. In this context, our data indicate that ascorbate could be a “Dr Jekyll and Mr Hyde”27 partner: (i) because ascorbate concentrations in the extracellular fluid in the brain are estimated to be 200–400 μM, the prooxidant regime is possible; (ii) in contrast, ascorbate concentrations in neurons are much higher (10 mM) leading to the antioxidant regime.3 However, even at much higher ascorbate concentration the antioxidant effect is incomplete as CCA can still trap HO˙. This raises the question whether it is possible to reach high enough ascorbate or other sacrificial anti-oxidant1 concentration in vivo, so that they could completely scavenge HO˙ produced by Cu(Aβ). Using compounds that can redox silence the Cu bound to Aβ (and hence stop ROS production in general) might be a more efficient strategy. Cu chelating agents that can bind and/or remove Cu ions from Aβ and redox silence them are among such compounds.28–31 Another possibility would be to use catalytic ROS scavengers, such as SOD mimics.32,33
While high ascorbate concentrations could have an antioxidant effect for other biomolecules, they cannot prevent Aβ from oxidative damage. Whether this would be a beneficial or detrimental process depends on the toxicity of the oxidized Aβ, which is not clear yet.
The authors acknowledge L. Debrauwer and E. Jamin for providing the Orbitrap mass spectrometer (MetaToul-AXIOM, INRA, UMR1331 Toxalim, Toulouse, France). F.C. and P. F. thank the ANR – ANR-13-BSV5-0016; C.H. thanks the ERC aLzINK – contract no. 638712 for financial support.
Notes and references
- F. Di Domenico, E. Barone, M. Perluigi and D. A. Butterfield, Expert Rev. Neurother., 2015, 15, 19 CrossRef CAS PubMed
.
- H. Sies, Eur. J. Biochem., 1993, 215, 213 CrossRef CAS PubMed
- M. E. Rice, Trends Neurosci., 2000, 23, 209 CrossRef CAS PubMed
- B. Halliwell, J. Neurochem., 2006, 97, 1634 CrossRef CAS PubMed
- G. R. Buettner and B. A. Jurkiewicz, Radiat. Res., 1996, 145, 532 CrossRef CAS PubMed
- I. D. Podmore, H. R. Griffiths, K. E. Herbert, N. Mistry, P. Mistry and J. Lunec, Nature, 1998, 392, 559 CrossRef CAS PubMed
- A. Carr and B. Frei, FASEB J., 1999, 13, 1007 CAS
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, J. Struct. Biol., 2006, 155, 30 CrossRef CAS PubMed
- C. Hureau, Coord. Chem. Rev., 2012, 256, 2164 CrossRef CAS
- D. E. Hartter and A. Barnea, Synapses, 1988, 2, 412 CrossRef CAS PubMed
- J. Kardos, I. Kovács, F. Hajós, M. Kálmán and M. Simonyi, Neurosci. Lett., 1989, 103, 139 CrossRef CAS PubMed
- D. J. Bonda, X. Wang, G. Perry, A. Nunomura, M. Tabaton, X. Zhu and M. A. Smith, Neuropharmacology, 2010, 59, 290 CrossRef CAS PubMed
- D. G. Smith, R. Cappai and K. J. Barnham, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1976 CrossRef CAS PubMed
- Y. Manevich, K. D. Held and J. E. Biaglow, Radiat. Res., 1997, 148, 580 CrossRef CAS PubMed
- C. Hureau and P. Faller, Biochimie, 2009, 91, 1212 CrossRef CAS PubMed
- S. Noël, F. Perez, J. T. Pedersen, B. Alies, S. Ladeira, S. Sayen, E. Guillon, E. Gras and C. Hureau, J. Inorg. Biochem., 2012, 117, 322 CrossRef PubMed
- B. Alies, I. Sasaki, O. Proux, S. Sayen, E. Guillon, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 1214 RSC
- E. Atrián-Blasco, E. Cerrada, A. Conte-Daban, D. Testemale, P. Faller, M. Laguna and C. Hureau, Metallomics, 2015, 7, 1229 RSC
- S. Chassaing, F. Collin, P. Dorlet, J. Gout, C. Hureau and P. Faller, Curr. Top. Med. Chem., 2013, 12, 2573 CrossRef
- T. Kowalik-Jankowska, M. Ruta, K. Wiśniewska, L. Łankiewicz and M. Dyba, J. Inorg. Biochem., 2004, 98, 940 CrossRef CAS PubMed
- R. C. Nadal, S. E. J. Rigby and J. H. Viles, Biochemistry, 2008, 47, 11653 CrossRef CAS PubMed
- V. Balland, C. Hureau and J.-M. Saveant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113 CrossRef CAS PubMed
- L.-E. Cassagnes, V. Hervé, F. Nepveu, C. Hureau, P. Faller and F. Collin, Angew. Chem., Int. Ed., 2013, 52, 11110 CrossRef CAS PubMed
- L. G. Trujano-Ortiz, F. J. González and L. Quintanar, Inorg. Chem., 2014, 54, 4 CrossRef PubMed
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensin, Coord. Chem. Rev., 2012, 256, 2129 CrossRef CAS
- T. Persson, B. O. Popescu and A. Cedazo-Minguez, Oxid. Med. Cell. Longevity, 2014, 2014 Search PubMed
-
R. L. Stevenson, Strange case of Dr Jekyll and Mr Hyde, Longmans, Green, and Co., London, 1886 Search PubMed
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 16, 67 CrossRef CAS PubMed
- C. Rodríguez-Rodríguez, M. Telpoukhovskaia and C. Orvig, Coord. Chem. Rev., 2012, 256, 2308 CrossRef
- K. J. Barnham and A. Bush, Chem. Soc. Rev., 2014, 43, 627 RSC
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475 CrossRef CAS PubMed
- I. Batinic-Haberle, A. Tovmasyan, E. R. Roberts, Z. Vujaskovic, K. W. Leong and I. Spasojevic, Antioxid. Redox Signaling, 2014, 20, 2372 CrossRef CAS PubMed
- I. Batinic-Haberle, S. Reboucas Julio and I. Spasojevic, Antioxid. Redox Signaling, 2010, 13, 877 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, conditions for HO˙ detection using CCA, and LC-MS quantification of the oxidized peptide. See DOI: 10.1039/c6dt01979j |
| ‡ Current address: Institut de Chimie (UMR 7177), 4 rue B. Pascal, F-67000 Strasbourg, France. E-mail: pfaller@unistra.fr |
| § For technical reasons, the Aβ28 peptide encompassing the first 28 amino-acid residues has been used in mass spectrometry experiments (see the ESI† for details). |
| ¶ Note that data shown in Fig. 2 and 3 are representative from a series of measurements performed under slightly different conditions, but for which the very same trends were observed. |
| This journal is © The Royal Society of Chemistry 2016 |