
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of Zr(IV)-based architectures generated from ligands incorporating the 2,2′-biphenolato unit†
Chengrui
Miao
,
Georges
Khalil
,
Alain
Chaumont
,
Pierre
Mobian
* and
Marc
Henry
Laboratoire de Chimie Moléculaire de l'Etat Solide (Chimie de la Matière Complexe, UMR 7140), Université de Strasbourg, 67070 Strasbourg, France. E-mail: mobian@unistra.fr
First published on 12th April 2016
Abstract
The structural identification in solution of the Zr(IV) complexes involving two 2,2-biphenol-based proligands is reported. The proligand L1H2 contains one 2,2-biphenol unit whereas L2H4 incorporates two 2,2-biphenol units linked by a para-phenylene bridge. Diffusion Ordered Spectroscopy (DOSY) combined with electrospray mass spectrometry analysis and density functional theory (DFT) allowed for determining the molecular structures of such Zr(IV)-based architectures. It is proposed that [Zr(OPri)4(HOPri)] in the presence of L1H2 generates an octahedral complex formulated as [ZrL13H2]. Concerning the self-assembled architecture incorporating the L2 ligand, the analytical data highlight the formation of an unprecedented neutral Zr(IV) triple-stranded helicate ([Zr2L23H4]). Insight into the geometry of these complexes is obtained via DFT calculations. Remarkably, the helicate structure characterized in solution strongly contrasts with the triple-stranded structure of the complex that crystallizes.
Metal alkoxides or aryloxides are a major class of compounds that are extensively employed as precursors for sol–gel chemistry and materials science.1 Their chemistry is recognized as being highly complex since the predictable synthesis of complexes starting from these metallic precursors is extremely challenging. Undoubtedly, this complex chemistry arises from the singular reactivity of metal alkoxide or aryloxide complexes. Metal alkoxide or aryloxide species display generally complex behaviours in solution owing to their strong tendencies for oligomerisation via the formation of oxo, hydroxo and/or alkoxo-bridges between metals leading to solid-state molecular structures that are frequently far from those expected for complexes in solution.2 Recently, we have demonstrated the possibility of incorporating biphenolate-Ti(IV) motifs in a predictable way into self-assembled molecular structures. The rational approach adopted by our group has permitted us to generate helical architectures starting from titanium isopropoxide and a zig-zag strand incorporating two 2,2′-biphenol units. In order to extend this metallo-supramolecular chemistry to other metal alkoxide precursors, we describe in this manuscript the coordination chemistry involving substituted 2,2′-biphenol ligands with the Zr(IV) centre.
In this manuscript we report the liquid state characterisation of the architectures resulting from the reactions of the Zr(IV) centre with 2,2′-biphenol-based compounds. The proligands L1H2 and L2H43 involved in this study are depicted in Fig. 1. As mentioned above, this set of 2,2′-biphenol derivatives has been shown to generate complex controlled assemblies in the presence of the Ti(IV) centre. For instance, L1H2 leads to a monomeric C2-symmetric complex constructed around an octahedral titanium centre. This compound is formulated as [Ti(L1)2(HOPri)2].4 Concerning the strand L2H4, helical neutral assemblies named [Ti3(L2)3(HOPri)6] and [Ti2(L2)(L2H)(OPri)(HOPri)2] were characterised.5 Having proved that these ligands are well suited for displaying a rich Ti(IV) coordination chemistry, we examine now the role of the metal ion size on the resulting assemblies. Thus, it appeared to us particularly relevant to study the coordination chemistry of these two compounds, i.e. L1H2 and L2H4 in the presence of zirconium(IV). Compared to Ti(IV), Zr(IV) is known for being bigger with a Pauling ionic radius ratio between Zr(IV) and Ti(IV) equal to r(Zr(IV))/r(Ti(IV)) = 1.15.6 Thus, Zr(IV)-complexes with coordination numbers higher than six are rather common in comparison with Ti(IV)-complexes.7 Also, the large ionic radius of Zr(IV) permits accommodation of bulky ligands around this metallic centre.8
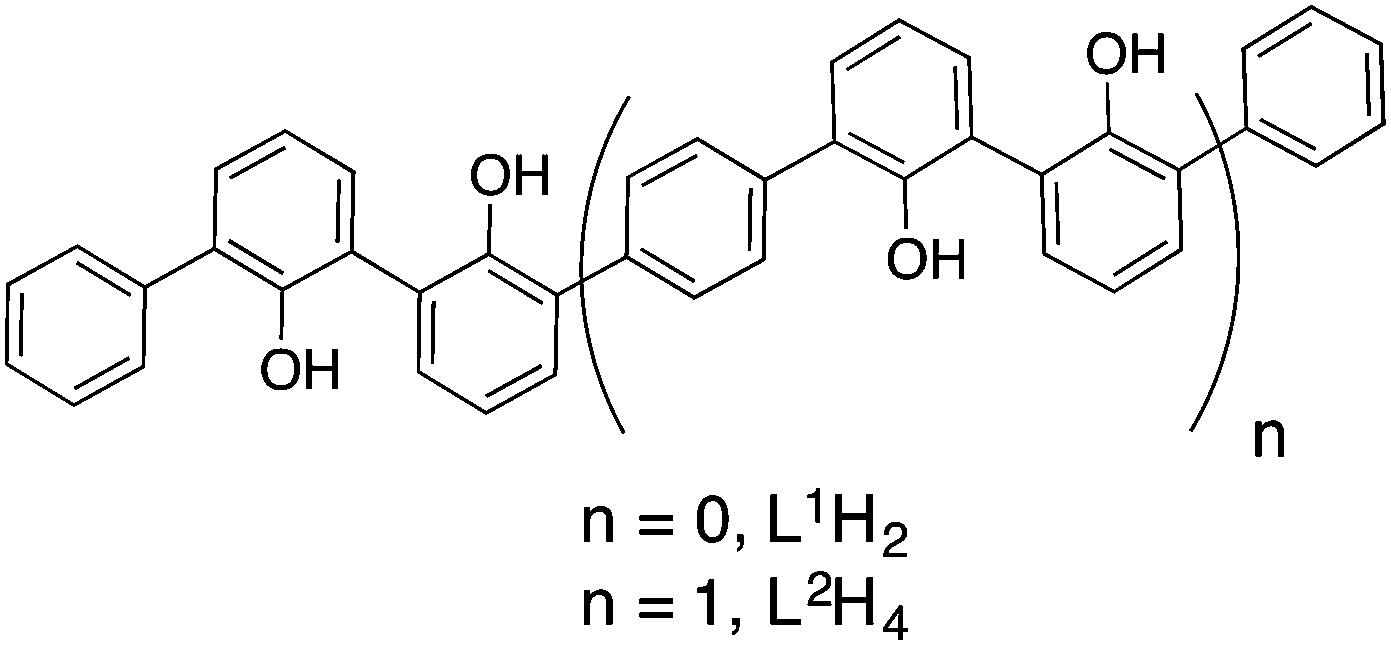 | ||
| Fig. 1 Compounds L1H2 and L2H4. | ||
Hereafter, we report the methodology allowing to elucidate the formula of the compounds resulting from the reactions of L1H2 or L2H4 with Zr(IV). We show that DOSY supported by mass spectrometry analysis and DFT calculations are a key combination to elucidate the nature of these species formed in situ. This study shows that the structures incorporating the Zr(IV) ions are dissimilar from those already mentioned for Ti(IV). In particular, this investigation highlights that Zr(IV) accommodates three bidentate oxygenated L1 ligands. This particular feature allows the formation of an assembly composed of two Zr(IV) centres and three strands when Zr(IV) reacts with L2H4. DFT calculations suggest for the dinuclear assembly a triple-stranded helicate structure formulated as Zr2L23H4. Finally, we show that this metallo-supramolecular chemistry conducted with Zr(IV) and L2H4 generates in the solid state an assembly strongly dissimilar from the one characterized in solution.
Results and discussion
ZrL13H2
The zirconium source employed to conduct this study was the “classical” [Zr(OPri)4(HOPri)] alkoxide complex. It is noteworthy that this species adopts a dimeric structure involving di-μ-isopropoxo bridges ([Zr2(OPri)8(HOPri)2]) in solution and in the solid state with two intramolecular hydrogen bonds characterized for this compound.9 First, several reaction conditions were tested by reacting [Zr(OPri)4(HOPri)] with 1, 2 and 3 equivalents of L1H2 in CD2Cl2. The resulting uncoloured mixtures were analysed by 1H NMR (see the ESI†). Surprisingly, these coordination reactions were not efficient as the L1H2 compound was largely predominant in solution for the three molar ratios tested. Also, the final composition of these mixtures was not really affected when these samples were heated. The most promising result was obtained when [Zr(OPri)4(HOPri)] reacted with two equivalents of L1H2. New large signals different from those observed for the initial components were clearly identified by 1H NMR as shown in Fig. 2, highlighting the formation of a Zr-based complex. A conversion ratio of about 51% could be estimated by 1H NMR after integration of the most deshielded signal of the unreacted L1H2 and the methine proton signal (δ = 3.93 ppm, broad signal). We explain this conversion by the dimeric structure of [Zr2(OPri)8(HOPri)2] rendering this precursor moderately reactive.1 The aliphatic region of the 1H NMR spectrum displays two intense very broad resonances at δ = 3.89 ppm and 1.09 ppm (see the ESI†). Overall, the shape of these signals suggests an exchange process with free alcohol generated in the course of the coordination step of L1H2 and the ligands of the dimeric [Zr2(OPri)8(HOPri)2] precursor.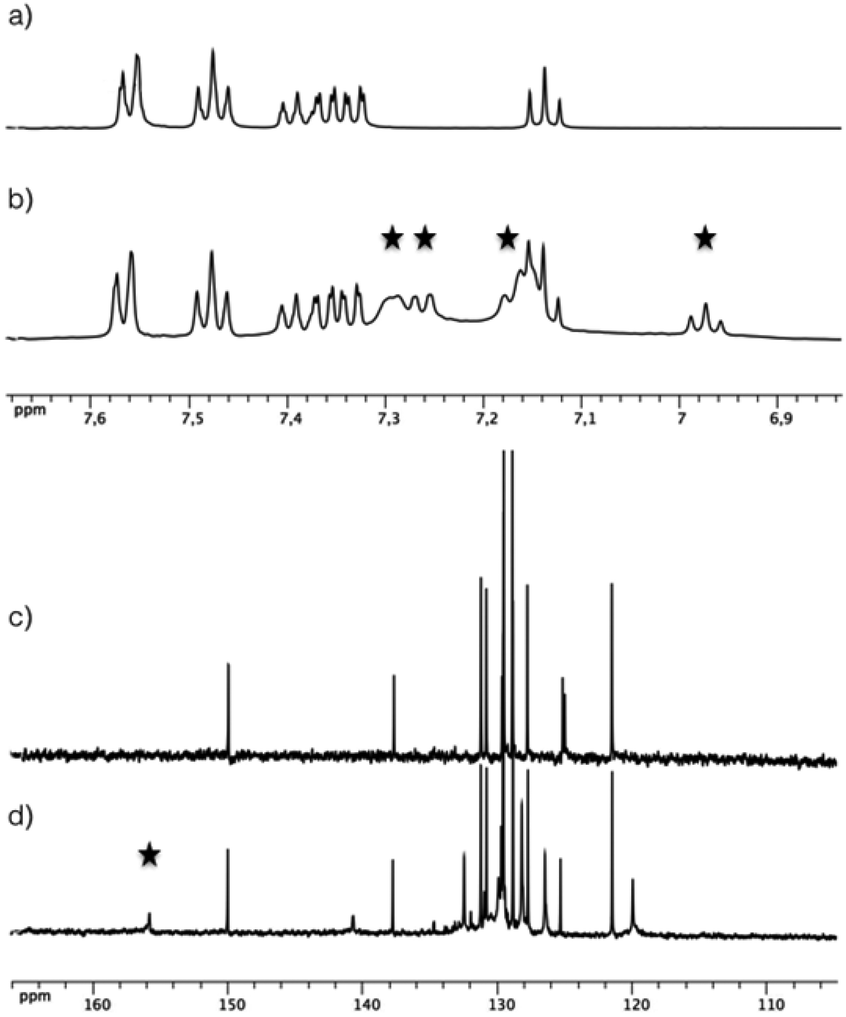 | ||
| Fig. 2 1H NMR (a) and 13C NMR (c) of L1H2 in CD2Cl2 (aromatic region). 1H NMR (b) and 13C NMR (d) of the mixture resulting from the reaction of [Zr(OPri)4(HOPri)] with two equivalents of L1H2 in CD2Cl2 (aromatic region). Signals marked with the star correspond to selected signals attributed to the complex formed in situ. | ||
A single deshielded signal (δ = 155.0 ppm) was observed by 13C NMR spectroscopy (Fig. 2c and d), in the region corresponding to a chemical shift typical to the ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –O–Zr resonance.10 Consequently, this 13C NMR spectrum is in full accordance with the formation of a single highly symmetrical complex in solution. It is worth noticing that this complex appears to be unstable in solution as the 1H NMR aromatic region spectrum of a mixture analysed after one day of its preparation reveals only the presence in solution of the initial L1H2 molecule.
–O–Zr resonance.10 Consequently, this 13C NMR spectrum is in full accordance with the formation of a single highly symmetrical complex in solution. It is worth noticing that this complex appears to be unstable in solution as the 1H NMR aromatic region spectrum of a mixture analysed after one day of its preparation reveals only the presence in solution of the initial L1H2 molecule.
Among all the available analytical techniques permitting to evaluate the nature of self-assembled architectures or coordination compounds, Diffusion Ordered Spectroscopy (DOSY) experiments11 are becoming increasingly popular to elucidate these metallo-supramolecular complexes. In the absence of structural data for a given species, this non-invasive technique permits to gain crucial information about the size and the shape of a compound in solution since the diffusion coefficients measured in the course of a DOSY experiment are correlated to the hydrodynamic radius (Rh) of the species in solution according to the Stokes–Einstein equation.12 Therefore, DOSY became a routine technique to analyse metallo-supramolecular aggregates in solution.13 For instance, this NMR method has been successfully applied to analyse helicates,14 cyclic inorganic clusters,15 cages,16 rotaxanes,17 catenanes18 or metallo-prisms.19 Thus, DOSY appeared to us as a well adapted technique to analyse the mixtures obtained when Zr(IV) reacts with L1H2 and later with L2H4. Table 1 gathers the diffusion coefficients measured for the species in solution and the diffusion coefficients measured for the previously reported Ti(IV)-based references. These references were synthesized by using analogous conditions as those reported here. For the experiment conducted with two equivalents of L1H2 and one equivalent of [Zr(OPri)4(HOPri)], only two species containing aromatic protons are detected by DOSY; L1H2 and a new species assumed for being a Zr(IV)-complex (see the ESI†). The diffusion measured for the complex corresponds only to aromatic proton resonances showing that the Zr(IV) ion is not coordinated to isopropanol or isopropoxo ligands. The complex formed in situ is around 4.7 times more voluminous than the L1H2 compound (estimated volumes: V(Zr(IV)-complex) = 1420 Å3, V(L1H2) = 300 Å3, where V is calculated from Rh values with the relation V = 4/3·π·Rh3 assuming a spherical shape of the molecules). A volume ratio of 1.5 is found between the Zr(IV)-based complex in solution and a previously reported complex with V(Zr(IV)-complex) = 1420 Å3 and V([Ti(L1)2(HOPri)2]) = 940 Å3 highlighting a Zr(IV)-complex containing three ligands. Finally, electrospray ionization mass spectrometry (ESI-MS) analysis allows the unambiguous identification of the complex. As shown in Fig. 3, two intense peaks are detected by ESI-MS. According to the peak observed at high m/z (m/z = 1123.2561), a formula [ZrL13H2] could be proposed for this Zr(IV)-based compound formed in situ.
 | ||
| Fig. 3 ESI mass spectrum of the mixture obtained after mixing two equivalents of L1H2 with [Zr(OPri)4(HOPri)] in CD2Cl2. The experimental peak (top) at m/z = 1123.2561 is shown in the insert picture with the isotopic profile (bottom) simulated for [ZrL13H2 + Na]+ (calcd for C72H50NaO6Zr; m/z = 1123.2547). The peak at m/z = 699.2470 is attributed to [2L1H2 + Na]+ (calcd for C48H36NaO4; m/z = 699.2511). | ||
| Complex in solution | Diffusion (μm2 s−1) | R h (Å) |
|---|---|---|
| a The following stoichiometry has been applied to generate the complex in situ: 2L1H2 + [Zr(OPri)4(HOPri)]. b The following stoichiometry has been applied to generate the complex in situ: L2H4 + [Zr(OPri)4(HOPri)]. | ||
| [ZrL13H2]a | 680 | 7.0 |
| [Zr2L23H4]b | 505 | 9.4 |
| References | ||
| L1H2 | 1150 | 4.1 |
| L2H4 | 830 | 5.7 |
| [Ti(L1)2(HOPri)2]4 | 780 | 6.1 |
[Ti2(L2)(L2H)(OPri)(HOPri)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 5 |
630 | 7.5 |
Zr2L23H4
Having determined the formula of the Zr(IV) complex formed in situ with L1H2, the reaction involving the L2H4 proligand was studied. In this case, L2H4 reacts with one equivalent of [Zr(OPri)4(HOPri)] at room temperature. Neither 1H NMR nor 13C NMR analysis permitted us to extract useful information about the species formed in situ. The 1H NMR spectrum indicates the presence of unreacted L2H4 in solution with also the presence of large and complex broad signals in the aromatic region too complex to be analysed (see the ESI†). Herein, the coordination process is rather efficient. A conversion of 87% was estimated by integrating the para-phenylene bridge proton signal of the unreacted L2H4 (δ = 7.68 ppm, singlet) and the methine proton signal (δ = 3.93 ppm, broad septuplet). The 13C NMR spectrum only displays signals relative to L2H4 in the region of aromatic carbons. By DOSY, in addition to the diffusion attributed to the unreacted L2H4 strand, only one diffusion associated to the formation of a Zr(IV)-based assembly incorporating the ligand L2 is noticed for the analysed solution. This observation highlights the high selectivity of the self-assembly process. Indeed in this particular case, the molecular volumes could not be estimated accurately for this species, as the shape of the complex in solution is unknown. However, owing to the order of magnitude of the Rh values (see Table 1), it appears that the Zr(IV)-based assembly is more voluminous than the titanium(IV) dinuclear double stranded helicate already reported.5 ESI-MS analysis of the crude mixture proves the formation of a dinuclear assembly built up from three strands. The ESI-MS mass spectrum and the simulated isotopic profile for the peak at m/z = 2006.4081 are shown respectively in Fig. 4a and b. The two more intense peaks at m/z = 1214.4673 and m/z = 1813.6810 are originated from LH4. Peaks at m/z = 1984.4188 and 2006.4081 are attributed to a species formed by two zirconium ions and three strands. In addition to these parent peaks, a peak at m/z = 1898.5531 is detected. This peak is originated from a monomeric complex bearing two strands partially protonated and one strand fully protonated ([ZrL23H8(H2O)]+). Now, the dinuclear architecture formed in situ could be assigned as Zr2L23H4. This result is in agreement with the conclusion made for the experiment conducted with the L1H2 ligand.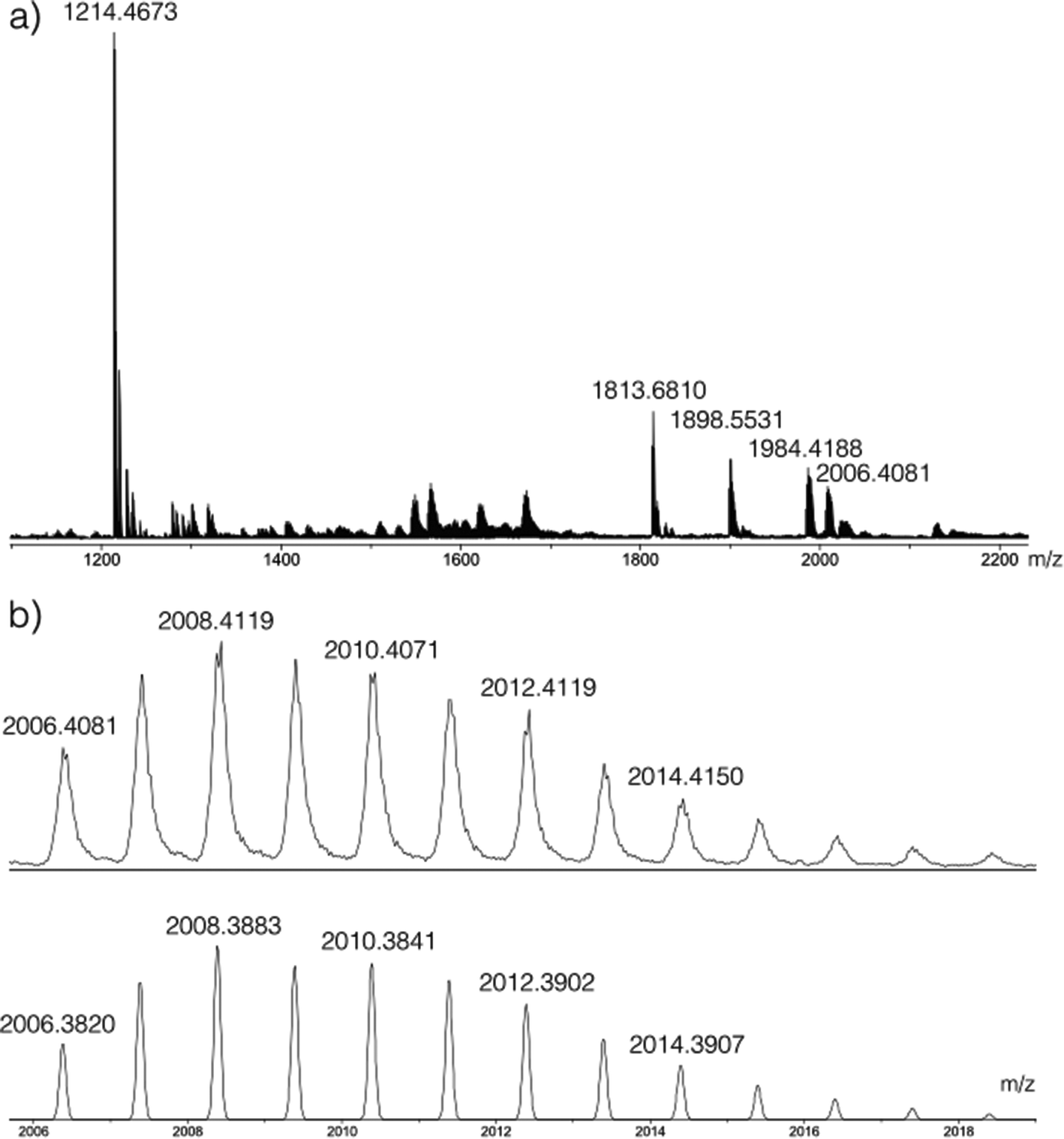 | ||
| Fig. 4 (a) ESI mass spectrum of the mixture obtained after reacting two equivalents of L2H4 with [Zr(OPri)4(HOPri)] in CD2Cl2. Peaks at m/z = 1214.4673 and 1813.6810 are attributed to L2H4 (calcd for [3L2H4 + H2O]+ = 1813.6566 and calcd for [2L2H4 + H2O]+ = 1214.4388). Peaks at m/z = 1984.4188 and 2006.4081 correspond to an assembly incorporating two Zr(IV) centres and three L2 ligands (calcd for [Zr2L23H4(H2O)]+ (C126H84O13Zr2) = 1984.4040 (see the ESI† for the isotopic simulation) and calcd for [Zr2L23H4(H2O)Na]+ (C126H83NaO13Zr2) = 2006.3825). The simulated isotopic profile for the C126H83NaO13Zr formula (bottom) and the enlargement of the peak (top) at m/z = 2006.4081 are given in (b). The peak at m/z = 1898.5531 is assigned to [ZrL23H8(H2O)]+ (calcd for C126H88O13Zr, m/z = 1898.5272) (simulated isotopic profile is given in the ESI†). | ||
Molecular modeling
In order to gain detailed information about the Zr(IV)-based architectures generated from L1H2 and L2H4, molecular models were computed and their energies minimized using density functional theory (DFT) with the hybrid exchange correlation functional B3LYP. The structures of the mononuclear and dinuclear complexes were modelled on the basis of the conclusion drawn by the DOSY experiments and the mass spectrometry analysis. The monomeric complex is designed knowing that the metallic centre is linked to three bidentate ligands. Owing to the reason that in this complex two phenol sub-units compose the Zr(IV) coordination sphere, the three structures [ZrL12L1H2], [cis-ZrL1(L1H)2] and [trans-ZrL1(L1H)2] were optimized (a schematic representation of these isomers is shown in Fig. 5). The calculated structure corresponding to [cis-ZrL1(L1H)2] is presented in Fig. 6, and the structures of [ZrL12L1H2] and [trans-ZrL1(L1H)2] are shown in the ESI.†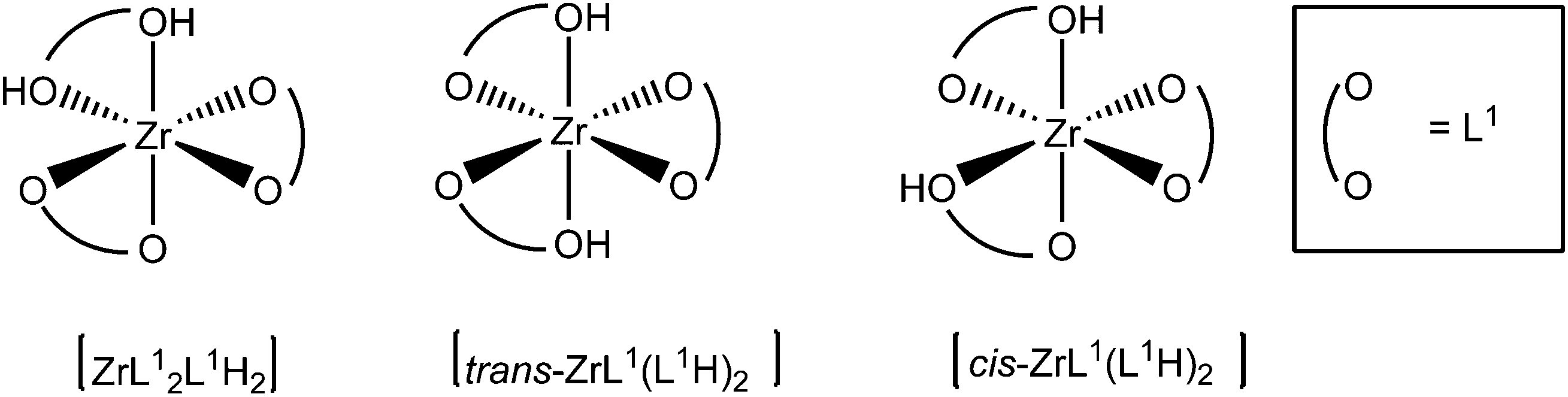 | ||
| Fig. 5 Schematic representation of the three isomers simulated for [ZrL13H2]. | ||
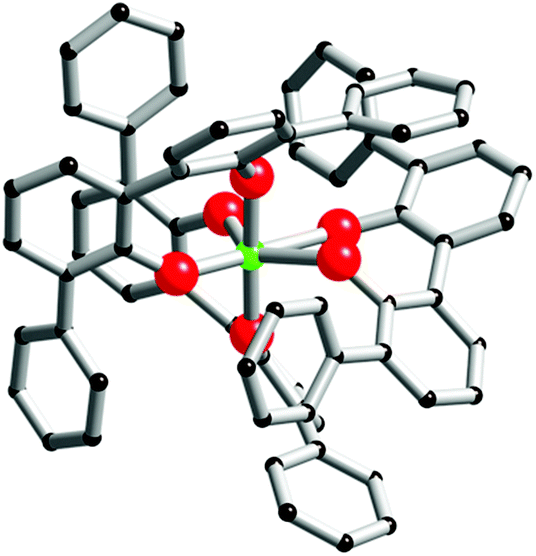 | ||
| Fig. 6 Computer model of [cis-ZrL1(L1H)2]. The zirconium atom is green, oxygen atoms are red and carbon atoms are black. For these two models, hydrogen atoms are omitted for clarity. | ||
The [cis-ZrL1(L1H)2] isomer appears to be the most stable structure. The energetic difference between [cis-ZrL1(L1H)2] and [trans-ZrL1(L1H)2] is found to be 39 kJ mol−1, while between [cis-ZrL1(L1H)2] and [ZrL12L1H2] an energy difference of 47 kJ mol−1 is determined. As shown in Fig. 6, the [cis-ZrL1(L1H)2] model indicates that the metallic centre is large enough to accommodate the three ligands leading to a metallic coordination sphere composed of six oxygen atoms. As expected the Zr–O bond lengths are not equivalent as Zr–O(phenolato) bonds (Zr–O(phenolato) = 2.01 ± 0.03 Å) are shorter comparatively to Zr–O(phenol) bonds (Zr–O(phenol) = 2.35 ± 0.005 Å). From the optimized structure, the Rh has been estimated with the PACHA software.20 The computed Rh determined for [cis-ZrL1(L1H)2] is the average between a maximum value corresponding to the longest distance from the barycentre of the molecule and a minimum value Rmin = (3V/4π)1/3 derived from the molecular volume21 (van der Waals radii used: H = 1.2 Å, C = 1.7 Å, O = 1.52 Å,22 and Zr = 2.00 Å (assumed)). The estimated Rh value (Rh = 7.56 ± 0.4 Å) is in good agreement with the one determined experimentally by DOSY for [ZrL13H2] (Rh = 7.00 ± 0.7 Å).
Next, the computed models incorporating the bis-2,2′-biphenolato strand were calculated. Herein, the modelled structures concern dinuclear species surrounded by three strands. These complexes display to be having the Zr2L22L2H4 formula. In these structures, we imposed the full protonation of one L2 strand. As two stereoisomers have to be considered when a dinuclear triple stranded architecture is discussed, the helicate and the mesocate structures23 were optimized. The models of these two isomers are shown in Fig. 7. The helicate is a much more stable architecture comparatively to the mesocate. An energy difference of 368 kJ mol−1 in favour of the Zr2L22L2H4 triple-stranded helicate was found. In the case of the mesocate, the ligand L2 appears to be not flexible enough to permit the formation of such an isomer without any constraint. This is illustrated by the angle of 149.63° for C74–C68–C59 measured in the computed structure, whereas an almost perfect alignment of these carbons is expected (carbons C74, C68, C59 are labelled in Fig. 7b). In the case of the helicate structure, the measured angles and distances are reasonable and do not reflect strong constraints for the three L ligands. For the [Zr2L22L2H4] helicate, an estimated intermetallic distance of 8.5 Å is measured. The three organic strands wrap around the two zirconium atoms leading to an ovoid shape for the molecule. As it is noticed for [cis-ZrL1(L1H)2], the Zr–O(phenol) bonds are much longer than the Zr–O(phenolato) bonds (Zr–O(phenol) = 2.35 ± 0.04 Å; Zr–O(phenolato) = 2.01 ± 0.04 Å). By using the same procedure as the one used for the theoretical Rh value of cis-[ZrL1(L1H)2], a computed hydrodynamic radius of Rh = 9.7 ± 0.5 Å is determined from the model of the [Zr2L22(L2H4)] helicate. Again, this computed radius is in good agreement with the one observed by DOSY for the complex in solution (Rh = 9.4 ± 0.9 Å).
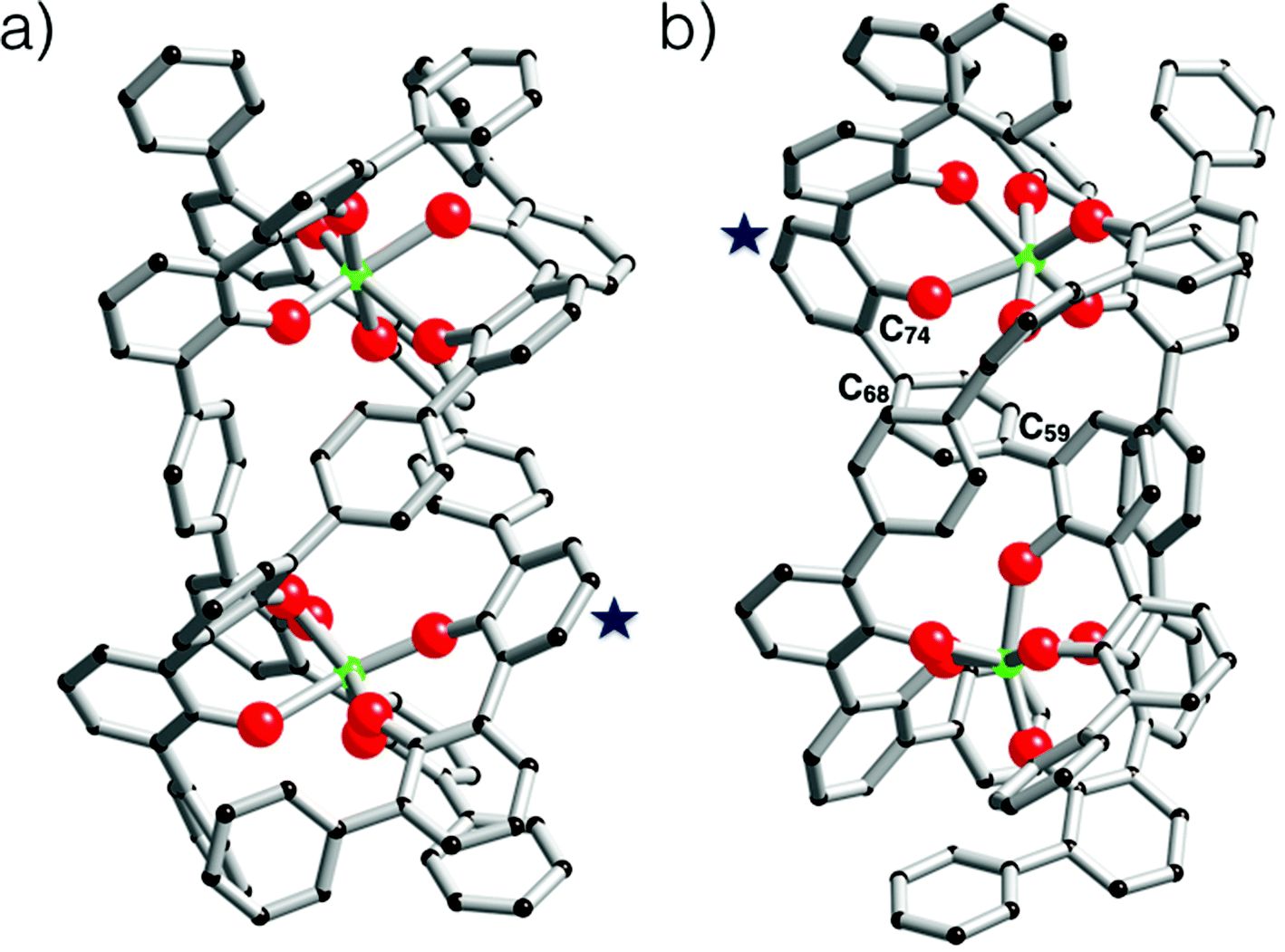 | ||
| Fig. 7 (a) Computer model of the [Zr2L22(L2H4)] helicate. In this model, one strand is fully protonated, and this strand is marked with a star. The computed structure represents the P-Δ, Δ enantiomer. (b) Computer model of the [Zr2L22(L2H4)] mesocate. In this model, one strand is fully protonated, this strand is marked with a star. This assembly contains a Δ and a Λ subunit. The angle measured between C74–C68–C59 is 149.63°. In these models four protons are linked to four oxygen atoms belonging to the same ligand. Zirconium(IV) atoms are green, oxygen atoms are red and carbon atoms are black. For these two models, hydrogen atoms are omitted for clarity. | ||
Crystal structure
Having evidenced the formation of defined architectures formed in situ when L1H2 or L2H4 react with [Zr(OPri)4(HOPri)], solid-state characterisation of these zirconium-based compounds was undertaken. Crystals suitable for X-ray diffraction analysis were obtained only in the case of ligand L2H4. Few white crystals were isolated after several weeks when vapours of diethylether were allowed to slowly diffuse in a dichloromethane solution composed of L2H4 (3 eq.) and [Zr(OPri)4(HOPri)] (2 eq.). The molecular structure obtained from X-ray diffraction analysis is shown in Fig. 8. The bond valence sum24 permitted addition of several missing H-atoms not located during refinement of the structure and the complex formula [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3] could be proposed. Importantly, this species was not detected by ES-MS in the course of the liquid state investigation. The [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3] complex incorporates three strands with one phenol group belonging to the L2H strand that is not coordinated to a zirconium centre. The three strands are wrapped around a zirconium hydroxo-ladder composed of four metallic centres. This particular arrangement leads to a helical architecture. Each zirconium centre is 6-fold coordinated with pseudo-octahedral coordination geometry as revealed by continuous shape measurements (Zr1(CShM) = 1.615; Zr2(CShM) = 2.756, Zr3(CShM) = 2.317; Zr4(CShM) = 1.1182).25 Four zirconium atoms form the inorganic core of the complex. The two central metallic atoms (Zr2 and Zr3) are μ2-hydroxo bridged by one another and are also respectively di-μ2-hydroxo bridged with a terminal zirconium(IV) centre (Zr1 or Zr4). Also, we notice that two and one isopropanol molecules coordinate with Zr1 and Zr4 respectively. It is noteworthy that the chain-like structure adopted by the inorganic core of the complex is rather original26 in comparison with the zirconium hydroxo- or oxo-clusters already reported. Here, the chain-like structure contrasts with the spherical or ovoid shapes of the inorganic cores usually found in zirconium-based aggregates.27 Undoubtedly, the chain-like structure found in [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3] is templated by the L2 strands.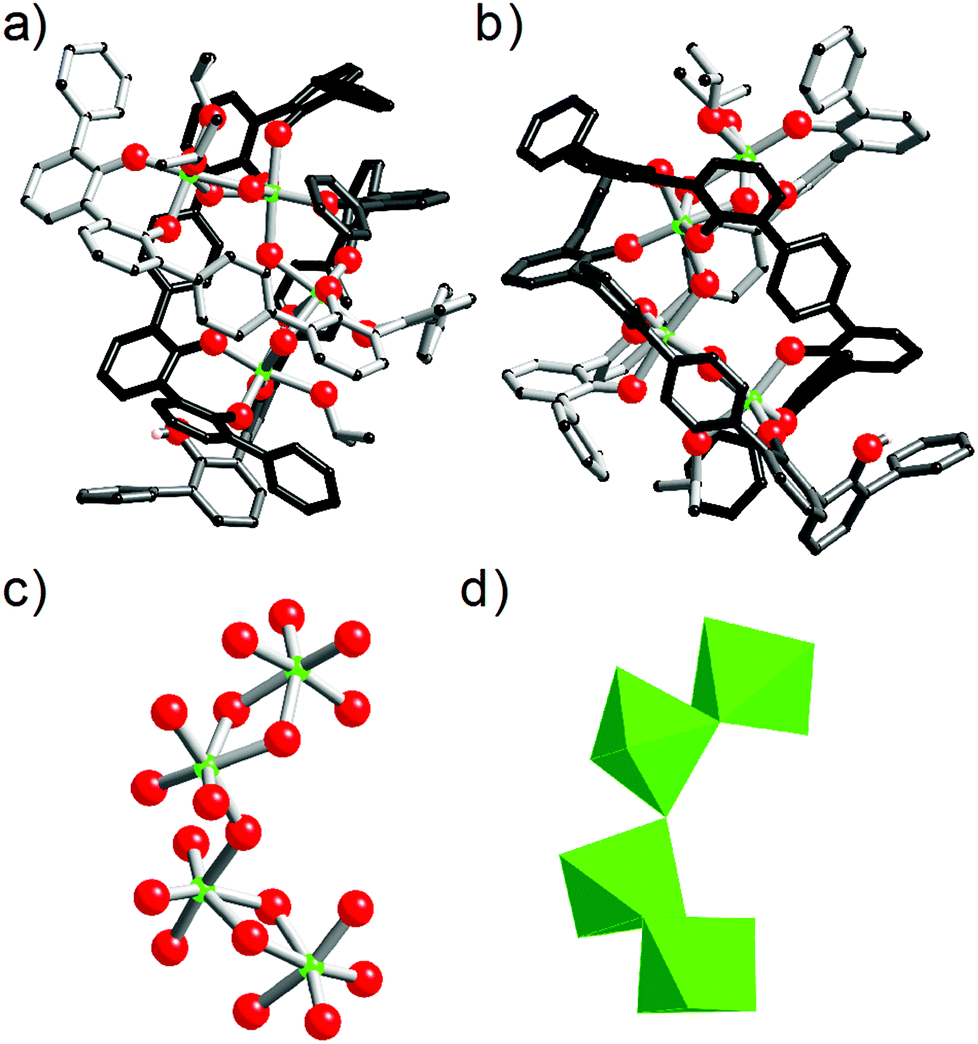 | ||
| Fig. 8 Two views (a) and (b) of the molecular structure of [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3]. In (c) is shown the inorganic core of the structure composed of four pseudo-octahedral ZrO6 units. (d) Polyhedral model of the inorganic core of the complex. For clarity the three strands are differently coloured and the hydrogen atoms are omitted with the exception of the hydrogen of L2H. Carbon atoms are in black, oxygen atoms in red and zirconium atoms in green. The disorder of several atoms is not shown. | ||
The structural difference observed between the structure formed in solution and the one observed in the solid-state is obviously related to the occurrence during crystallization of a partial hydrolysis of some Zr–OPh bonds followed by a classical olation reaction leading to Zr(μ-OH)2Zr bridges. As the occurrence of a [Zr2(L2)2(L2H4)] helicate in solution cannot be doubted, the first step could be the hydrolysis of two Zr–OPh bonds allowing the formation of the classical di-μ2-OH bridge causing departure of the fully protonated ligand (L2H4) with addition of two alcohol molecules in order to conserve the sixfold coordination of Zr-atoms:

The next step could be a further partial hydrolysis of the (L2H2) ligand according to:
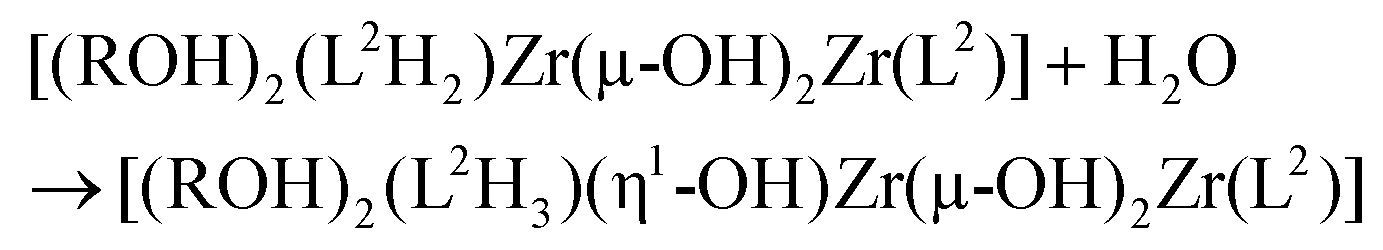
The solid-state structure could now be obtained through a single olation reaction between one (L2H2)-based and one (L2H3)-based hydrolysed dimers assisted by proton transfer between the (L2H2) and (L2H3) moieties in order to allow for departure of another neutral L2H4 ligand:
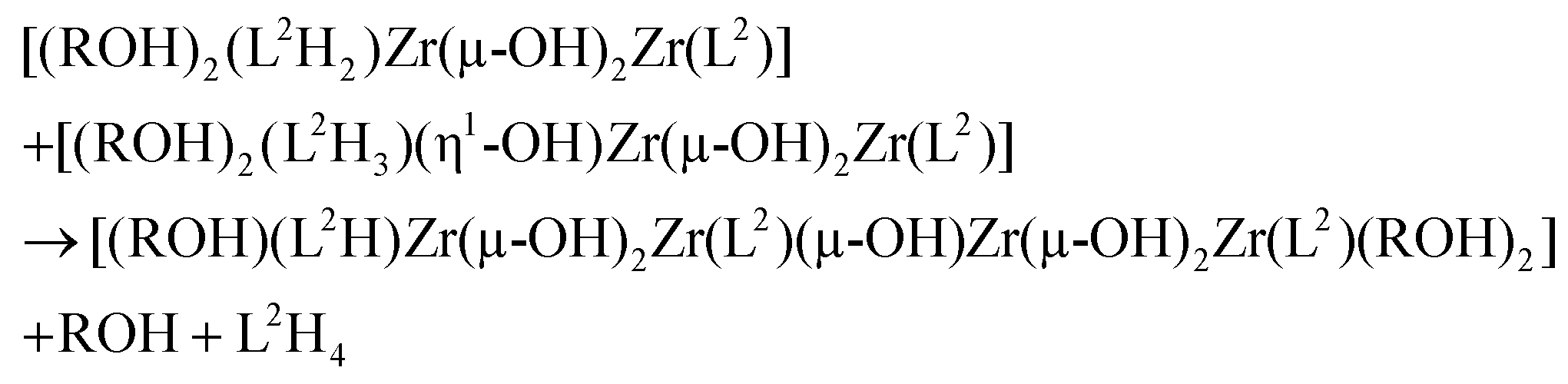
It is worth noticing that this filiation between the solution and the solid-state following the well-established general rules of sol–gel chemistry28 is in agreement with the following stoichiometric balance:
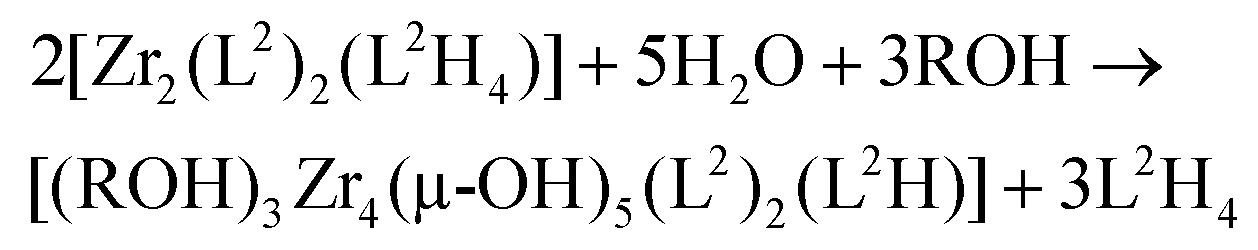
One may thus hope that under strictly anhydrous conditions, it could be possible to crystallize the [Zr2(L2)2(L2H4)] helicate.
From the structural viewpoint, this scheme supposes a large flexibility of the intermediates, a point that can only be addressed after careful molecular dynamics (MD) studies. However, in the absence of reliable MD potential for zirconium atoms, we have not been able to check this important point. The proposed stoichiometric condensation scheme should thus only be considered as a starting point for full modelling of such a complex condensation process. Another possibility would be that this helicate is indeed a strong acid that could be formulated as a negatively charged [Zr2(L2)3]4− helicate with 4 protons acting as counter-ions and delocalized over the whole structure. These “free” protons would then be able to catalyse the slow condensation of two alcohol molecules with the liberation of one water molecule: ROH + HOR → R–O–R + H2O. In such a case, the water necessary for the formation of the tetramer would be formed in situ preventing the isolation of the helicate in the crystalline state. Further theoretical work is thus needed in order to clarify this point.
Conclusion
We have described herein a coordination chemistry involving one of the most employed zirconium alkoxide precursors, i.e. [Zr(OPri)4(HOPri)], and two proligands incorporating the 2,2′-biphenol entities. The DOSY investigation supported by mass spectrometry and DFT calculations allowed us to suggest highly plausible structures for the complexes formed in situ. The coordination reaction involving the L1H2 proligand with Zr(IV) is less efficient comparatively to the one performed with Ti(IV). Nevertheless, we have established that the Zr(IV)-based complex formed from L1H2 incorporates three bidentate entities according to DOSY and ESI-MS analysis. DFT calculations indicate the formation of the [cis-ZrL1(L1H)2] isomer among three possible stereoisomers. Concerning the self-assembled architecture derived from the strand L2H4, the DOSY and mass spectrometry analysis proved the formation of a dinuclear assembly formed with three strands. Remarkably, computational modelling proposes an unprecedented triple-stranded helicate structure that incorporates two distorted octahedral ZrO6 units for the Zr(IV)-assembly. This structure evidence for the complex in solution contrasts strongly with the one obtained for the complex upon crystallization. In this case, a helical architecture constructed around an original chain-like inorganic core displaying the [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3] formula is characterized. It has been proposed that [Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3] may in fact be formed during crystallization owing to a partial hydrolysis/condensation reaction of the dimeric helicates mediated by water molecules.Overall, we have demonstrated the deep impact of the ionic size on the resulting self-assembled architecture formed with the strand L2 as with Ti(IV) only double-stranded helicates were obtained. Meanwhile, we have shown that the combination of Zr(IV) with the 2,2′-biphenol-based proligands leads to a less effective coordination chemistry to create stable and easily characterized self-assemblies in comparison with the one already developed with Ti(IV).
Experimental part
NMR spectroscopy
Bruker Avance-300, Avance-400 and Avance-500 were used for solution NMR spectroscopic analysis. 1H NMR DOSY measurements were performed at 500.13 MHz with a 5 mm 1H/X z-gradient BBI probe and by applying a PFGSTE pulse sequence using bipolar gradients. DOSY spectra were generated with the DOSY module of NMRNotebook™ software, through maximum entropy and inverse Laplace transform calculations. 1H NMR DOSY measurements were performed at 500.13 MHz with a 5 mm 1H/X z-gradient BBI probe and by applying a PFGSTE pulse sequence using bipolar gradients. The solutions were analysed using a NMR microtube. The viscosity of CD2Cl2 at 298 K was determined according ref. 5 (see the ESI†). Mass spectrometry was performed at the Service Commun d'Analyses, Université de Strasbourg (France). The electrospray analyses were performed on Micro-TOF (Bruker) apparatus equipped with an electrospray (ES) source. All solvents were dried before use with activated 3 Å molecular sieves. [Zr(OPri)4(HOPri)] was purchased from Strem. Ligands were synthesised according to ref. 3. All reactions were performed in a glove bag.X-ray diffraction
Data were collected on a Bruker SMART CCD diffractometer with Mo Kα radiation. The structures were solved using SHELXS-97 and refined by full matrix least-squares on F2 using SHELXS-97 with anisotropic thermal parameters for all nonhydrogen atoms. The hydrogen atoms were introduced at calculated positions and not refined (riding model).Computational details
Density functional theory (DFT) calculations on the different complexes have been performed with the Gaussian09 Revision D01 package.29 Hybrid exchange correlation functional B3LYP using the LANL2DZ basis set30–33 with an ultrafine grid was used in all our calculations. Geometry optimization of the [ZrL13H2] and [Zr2L23H4] complexes started from the crystallographic geometry from either the [Ti(L1)2(HOPri)2] (ref. 4) or [Ti2(L2)(L2H)(OPri)(HOPri)2] (ref. 5) complexes, where Ti atoms have been replaced by Zr atoms and to which an additional ligand was added. The minimum character of each stationary point was verified by computation of the harmonic vibrational frequencies, which were all real.[ZrL13H2]
The reaction was conducted under a nitrogen atmosphere. In a vial, L1H2 (10 mg, 0.03 mmol), and [Zr(OPri)4(HOPri)] (6 mg, 0.015 mol) were mixed in dry CD2Cl2 (0.75 mL). The resulting uncoloured mixture was analysed by 1H NMR, 13C NMR, DOSY and mass spectrometry. Some signals of the NMR spectrum were attributed to ZrL13H2: 1H NMR (CD2Cl2, 500 MHz): δ (ppm) = 6.97 (t, 7.5 Hz), 7.26 (bd dd, 4J = 1.5 Hz, 3J = 7.5 Hz), 7.29 (bd d); 13C NMR (CD2Cl2, 125 MHz): δ (ppm) = 155.8, 140.7, 132.4, 131.9, 130.9, 129.9, 128.2, 126.4, 119.9. ES-MS: m/z = 1123.2561 [ZrL13H2 + Na]+ (calcd for C72H50NaO6Zr; m/z = 1123.2547). DOSY: D = 680 μm2 s−1.[Zr2L23H4]
The reaction was conducted under a nitrogen atmosphere. In a vial, L2H4 (10 mg, 0.016 mmol), and [Zr(OPri)4(HOPri)] (6.45 mg, 0.016 mmol) were mixed in dry CD2Cl2 (0.75 mL). The resulting uncoloured mixture was analysed by 1H NMR, 13C NMR, DOSY and mass spectrometry. 1H NMR was too complex to be analysed. ES-MS: m/z = 2006.4081 [Zr2L23H4(H2O)]+ (calcd for C126H83NaO13Zr = 2006.3825). DOSY: D = 505 μm2 s−1.[Zr4(L2)2(L2H)(μ2-OH)5(HOiPr)3]
The reaction was conducted under a nitrogen atmosphere. Few colourless single crystals were obtained after several weeks by slow vapour diffusion of diethylether on a dry CH2Cl2 solution (1.5 mL) composed of L2H4 (15 mg, 2.5 10−5 mol), and [Zr(OPri)4(HOPri)] (6.5 mg, 0.66 eq.).X-ray data: empirical formula: C285H244Cl6O45Zr8 (2(C135H100O20Zr4), 3(C4H10O), 3(CH2Cl2), 2(H2O)′); formula mass: 5331.25 g mol−1; crystal system: triclinic; space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; unit cell dimensions: a = 17.0968(8) Å, b = 19.1728(8) Å, c = 21.3385(8) Å; α = 81.245(2)°, β = 68.379(2)°, γ = 87.843(2)°, V = 6425.4(5) Å3; Z = 1; density (calcd): 1.378 g cm−3; crystal size: 0.050 × 0.060 × 0.060 mm3; θ range for data collection: 1.28 to 30.15°; reflections collected: 33659; independent reflections: 33659 [R(int) = 0.0638]; refinement method: full-matrix least squares on F2; data/restraints/parameters: 33659/23/1565; goodness-of-fit on F2: 1.009; final R indices [I > 2σ(I)]: R1 = 0.0791, wR2 = 0.2240; R indices (all data): R1 = 0.1358, wR2 = 0.2446. CCDC number: 1451432.
; unit cell dimensions: a = 17.0968(8) Å, b = 19.1728(8) Å, c = 21.3385(8) Å; α = 81.245(2)°, β = 68.379(2)°, γ = 87.843(2)°, V = 6425.4(5) Å3; Z = 1; density (calcd): 1.378 g cm−3; crystal size: 0.050 × 0.060 × 0.060 mm3; θ range for data collection: 1.28 to 30.15°; reflections collected: 33659; independent reflections: 33659 [R(int) = 0.0638]; refinement method: full-matrix least squares on F2; data/restraints/parameters: 33659/23/1565; goodness-of-fit on F2: 1.009; final R indices [I > 2σ(I)]: R1 = 0.0791, wR2 = 0.2240; R indices (all data): R1 = 0.1358, wR2 = 0.2446. CCDC number: 1451432.
Acknowledgements
This work was done at Université de Strasbourg with public funds allocated by the CNRS, the French government and by the International Center for Frontier Research in Chemistry (icFRC scholarship to G. K.). We thank Dr L. Barloy for fruitful discussions. A. C. is grateful to the HPC centre of the Université de Strasbourg for computer resources. We thank N. Kyritsakas for X-ray structure resolution.References
- V. G. Kessler, in The Sol-Gel Handbook: Synthesis, Characterization and Applications, 3-Volume Set, John Wiley & Sons, 2015 Search PubMed; D. C. Bradley, R. Mehrotra, I. Rothwell and A. Singh, in Alkoxo and Aryloxo Derivatives of Metals, Academic Press, San Diego, 2001 Search PubMed; Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016 10.1039/C5CS00837A; F. Girardi, L. Fambri, S. Maggini and R. Di Maggio, J. Appl. Polym. Sci., 2015, 41568 Search PubMed; S. Gross, J. Mater. Chem., 2011, 21, 15853 RSC; T. J. Boyle, L. A. M. Ottley, S. M. Hoppe and C. F. Campana, Inorg. Chem., 2010, 49, 10798 CrossRef CAS PubMed; G. I. Spijksma, G. A. Seisenbaeva, A. Fischer, H. J. M. Bouwmeester, D. H. A. Blank and V. G. Kessler, J. Sol-Gel Sci. Technol., 2009, 51, 10 CrossRef.
- R. C. Mehrotra and A. Singh, Recent Trends in Metal Alkoxide Chemistry, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1997 Search PubMed.
- C. Diebold, D. M. Weekes, M. T. Navarrete, P. Mobian, N. Kyritsakas and M. Henry, Eur. J. Org. Chem., 2010, 6949 CrossRef CAS.
- C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Dalton Trans., 2009, 46, 10178 RSC.
- D. M. Weekes, C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Chem. – Eur. J., 2014, 20, 5092 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- L. Cooper, N. Guillou, C. Martineau, E. Elkaim, F. Taulelle, C. Serre and T. Devic, Eur. J. Inorg. Chem., 2014, 36, 6281 CrossRef CAS; H.-J. Chuang and B.-T. Ko, Dalton Trans., 2015, 44, 598 RSC; N. H. Lee, D. W. Lee, H. Yeo, K. Kwak, H. Sook Chun and K. M. Ok, CrystEngComm, 2014, 16, 5619 RSC; M. Steyn, H. G. Visser, A. Roodt and T. J. Muller, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1240–m1241 Search PubMed.
- A. Stopper, K. Press, J. Okuda, I. Goldberg and M. Kol, Inorg. Chem., 2014, 53, 9140 CrossRef CAS PubMed; M. Sietzen, H. Wadepohl and J. Ballmann, Organometallics, 2014, 33, 612 CrossRef; E. Otten, P. Dijkstra, C. Visser, A. Meetsma and B. Hessen, Organometallics, 2005, 24, 4374 CrossRef; M. P. Gil and O. L. Casagrande, J. Organomet. Chem., 2004, 689, 286 CrossRef; J. D. Scollard, D. H. McConville and J. J. Vittal, Organometallics, 1995, 14, 5478 CrossRef; E. Y. Tshuva, I. Goldberg and M. Kol, Organometallics, 2001, 20, 3017 CrossRef.
- B. A. Vaartstra, J. C. Huffman, P. S. Gradeff, L. G. Hubert-Pfalzgraf, J.-C. Daran, S. Parraud, K. Yunlu and K. G. Caulto, Inorg. Chem., 1990, 29, 3136 CrossRef.
- L. Azor, C. Bailly, L. Brelot, M. Henry, P. Mobian and S. Dagorne, Inorg. Chem., 2012, 10876 CrossRef CAS PubMed.
- E. L. Hahn, Phys. Rev., 1950, 80, 580 CrossRef; O. E. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288 CrossRef.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge University Press, Cambridge, 1984 Search PubMed.
- For a general review concerning the applications of DOSY in the field of supramolecular chemistry see: Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS PubMed.
- L. Allouche, A. Marquis and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 7520 CrossRef CAS PubMed.
- S. Floquet, S. Brun, J.-F. Lemonnier, M. Henry, A. Delsuc, Y. Prigent, E. Cadot and F. Taulelle, J. Am. Chem. Soc., 2009, 131, 17255 CrossRef PubMed.
- S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef CAS PubMed; S. Durot, L. Flamigni, J. Taesch, T. T. Dang, V. Heitz and B. Ventura, Chem. – Eur. J., 2014, 20, 9979 CrossRef PubMed; K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656 CrossRef PubMed.
- J. Frey, C. Tock, J.-P. Collin, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 2008, 130, 4592 CrossRef CAS PubMed.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950 CrossRef CAS PubMed.
- T. K. Ronson, J. Fisher, L. P. Harding and M. J. Hardie, Angew. Chem., Int. Ed., 2007, 46, 9086 CrossRef CAS PubMed.
- M. Henry, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, 2011, vol. 14, pp. 1–43 CrossRef CAS; H. Senouci, B. Millet, C. Volkringer, C. Huguenard, F. Taulelle and M. Henry, C. R. Chim., 2010, 13, 69 CrossRef CAS; M. Henry, in Advances in Quantum Chemical Bonding Structures, ed. M. V. Putz, Transworld Research Network, Kerala, India, 2009, pp. 153–211 RSC; C. Carpanese, S. Ferlay, N. Kyritsakas, M. Henry and M. W. Hosseini, Chem. Commun., 2009, 6786 RSC; M. Henry and M. W. Hosseini, New J. Chem., 2004, 28, 897 RSC; M. Henry, ChemPhysChem, 2002, 3, 561 CrossRef PubMed; M. Henry, ChemPhysChem, 2002, 3, 607 CrossRef PubMed.
- A. Gavezotti, J. Am. Chem. Soc., 1983, 105, 5220 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- For general reviews on helicates see: M. Albrecht, Chem. Rev., 2001, 101, 3457 CrossRef CAS PubMed; C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 CrossRef PubMed . For recent examples of triple-stranded helicates see: Q. Li, F. Huang, Y. Fan, Y. Wang, J. Li, Y. He and H. Jiang, Eur. J. Inorg. Chem., 2014, 20, 3235 CrossRef; C.-S. Tsanga, C.-C. Yeea, S.-M. Yiua, W.-T. Wong and H.-L. Kwong, Polyhedron, 2014, 83, 167 CrossRef; Q. Li, F. Huang, Y. Fan, Y. Wang, J. Li, Y. He and H. Jiang, Eur. J. Inorg. Chem., 2014, 20, 3235 CrossRef; O. Gidron, M.-O. Ebert, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2014, 53, 13614 CrossRef PubMed; C. Gütz, R. Hovorka, N. Struch, J. Bunzen, G. Meyer-Eppler, Z.-W. Qu, S. Grimme, F. Topić, K. Rissanen, M. Cetina, M. Engeser and A. Lützen, J. Am. Chem. Soc., 2014, 136, 11830 CrossRef PubMed; Z. Zhang and D. Dolphin, Chem. Commun., 2009, 6931 RSC.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1985, 41, 244 CrossRef.
- D. Casanova, M. Llunell, P. Alemany and S. Alavarez, Chem. – Eur. J., 2005, 11, 1479 CrossRef CAS PubMed.
- Few zirconium-based aggregates present a chain-like structure: B. Moraru, S. Gross, G. Kickelbick, G. Trimmel and U. Schubert, Monatsh. Chem., 2001, 132, 993 CrossRef CAS; T. J. Boyle, L. A. M. Ottley and M. A. Rodriguez, Polyhedron, 2005, 24, 1727 CrossRef.
- For zirconium-based aggregates presenting spherical or ovoid inorganic cores see: S. Yuan, W. Lu, Y.-P. Chen, Q. Zhang, T.-F. Liu, D. Feng, X. Wang, J. Qin and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 3177 CrossRef CAS PubMed; C. Artner, M. Czakler and U. Schubert, Inorg. Chim. Acta, 2015, 432, 208 CrossRef; V. Bon, I. Senkovska, M. S. Weiss and S. Kaskel, CrystEngComm, 2013, 15, 9572 RSC; P. Piszczek, A. Radtke, A. Grodzicki, A. Wojtczak and J. Chojnacki, Polyhedron, 2007, 26, 679 CrossRef; F. R. Kogler, M. Jupa, M. Puchberger and U. Schubert, J. Mater. Chem., 2004, 14, 3133 RSC; G. Kickelbick, P. Wiede and U. Schubert, Inorg. Chim. Acta, 1999, 284, 1 CrossRef; A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sánchez, L. Sánchez-Barba, M. Fernández-López and I. López-Solera, Inorg. Chem., 2004, 43, 1350 CrossRef PubMed; M. Y. Reza, H. Matsushima, M. Koikawa, M. Nakashima and T. Tokii, Polyhedron, 1999, 18, 787 CrossRef.
- C. Sanchez, J. Livage and M. Henry, Prog. Solid State Chem., 1988, 18, 259 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- T. H. Dunning Jr. and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1451432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00471g |
| This journal is © The Royal Society of Chemistry 2016 |