
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and testing of ZnO nanoparticles for photo-initiation: experimental observation of two different non-migration initiators for bulk polymerization†
M.
Schmitt
Saarland University, Campus B 2 2, 66123 Saarbruecken, Germany. E-mail: mic.schmitt@mx.uni-saarland.de
First published on 28th April 2015
Abstract
The migration and transport of polymerization initiators are problematic for commercially used polymerization procedures. For example, UV printing of packaging generates products with potentially harmful components that come in contact with food. Enlarging the size of the initiator is the only way to prevent contamination, e.g., by gas phase transport. In this manuscript, the synthesis and advanced and full analyses of novel nanoparticle-based types of non-migration, fragmenting and non-fragmenting photo-initiators will be presented in detail. This study introduces non-fragmenting/“Norrish type II” and fragmenting/“Norrish type I” ZnO nanoparticle-based initiators and compares them with two commercial products, a “Norrish type I” initiator and a “Norrish type II” initiator. Therefore, inter alia, the recently developed analysis involves examining the solidification by UV-vis and the double bond content by Raman. Irradiation is performed using absolute and spectrally calibrated xenon flash lights. A novel procedure for absolute and spectral calibration of such light sources is also presented. The non-optimized “Norrish type II” particle-based initiator is already many times faster than benzophenone, which is a molecular initiator of the same non-fragmenting type. This experimentally observed difference in reactive particle-based systems without co-initiators is unexpected. Co-initiators are normally an additional molecular species, which leads to migration problems. The discovery of significant initiation potential resulting in a very well-dispersed organic–inorganic hybrid material suggests a new field of research opportunities at the interface of physical chemistry, polymer chemistry and engineering science, with enormous value for human health.
Introduction
Polymerizations of monomeric units occur in nature and in an enormous number of man-made processes. Photo-induced bulk polymerization is used for material synthesis (e.g., polymers and coatings) in science and industry. UV curing is used in dental applications, artificial fingernail treatments and the printing and packing industries. In terms of economic and ecological considerations, UV printing is superior to any other solvent-based processes. Some advantages include high image quality, fast and comparatively easy production of printing plates, fast printing (e.g., more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sheets per h), the absence of additional solvents, and the low amount of monomer required due to film thicknesses of less than 2 μm. However, in applications such as food packaging, the remaining reacted initiator, which is necessary to solidify the resin, can be a severe problem as a result of migration. Migration is relevant for every application using molecular initiators. Multiple references concerning the problem of migration can be found on the World Wide Web and in the public press.1–10 The migration of initiators can be defined as a process resulting in the contamination of packaged contents by the initiation molecules or reaction products. For printing applications, the migration of initiators occurs by set-off (transfer from one sheet to the inner side of a second sheet), by diffusion and by gas-phase transport. The curing of films requires 3 to 5 wt% of initiators to overcome oxygen inhibition effects,11 whereby only a small concentration of the photo-initiator will be consumed. For example, initiators contents of up to 15 wt% are used for the flexo or UV printing of pigmented resins. Unfortunately, most well-known photo-initiators such as Irgacure®2959,12 Genocure®DMHA13 or Irgacure®MBF14 derive their chromophoric nature from aromatic functionalities, and most of them are small molecules. The highly reactive and mobile fragmentation products, daughter products and the remaining un-consumed initiators can pose a health threat.2,4 To prevent migration, the mass of the initiator can be increased by fabricating a polymeric initiator.13,15 Conversely, the danger can be reduced by combining a non-fragmenting “Norrish type II” initiator (Scheme 1) with the necessary co-initiator.16 The co-initiator is also sometimes called a sensitizer.13 The aim of this publication is to present novel alternative nanoparticle initiators, with the goal of preventing migration and as an innovative method of photo-polymerization. The use of a nano-initiator also produces very well-dispersed hybrid materials. Depending on their bandgap, semiconductors such as ZnO have the potential to absorb UV-light-forming excitons (electron–hole-pairs) (Scheme 1).
000 sheets per h), the absence of additional solvents, and the low amount of monomer required due to film thicknesses of less than 2 μm. However, in applications such as food packaging, the remaining reacted initiator, which is necessary to solidify the resin, can be a severe problem as a result of migration. Migration is relevant for every application using molecular initiators. Multiple references concerning the problem of migration can be found on the World Wide Web and in the public press.1–10 The migration of initiators can be defined as a process resulting in the contamination of packaged contents by the initiation molecules or reaction products. For printing applications, the migration of initiators occurs by set-off (transfer from one sheet to the inner side of a second sheet), by diffusion and by gas-phase transport. The curing of films requires 3 to 5 wt% of initiators to overcome oxygen inhibition effects,11 whereby only a small concentration of the photo-initiator will be consumed. For example, initiators contents of up to 15 wt% are used for the flexo or UV printing of pigmented resins. Unfortunately, most well-known photo-initiators such as Irgacure®2959,12 Genocure®DMHA13 or Irgacure®MBF14 derive their chromophoric nature from aromatic functionalities, and most of them are small molecules. The highly reactive and mobile fragmentation products, daughter products and the remaining un-consumed initiators can pose a health threat.2,4 To prevent migration, the mass of the initiator can be increased by fabricating a polymeric initiator.13,15 Conversely, the danger can be reduced by combining a non-fragmenting “Norrish type II” initiator (Scheme 1) with the necessary co-initiator.16 The co-initiator is also sometimes called a sensitizer.13 The aim of this publication is to present novel alternative nanoparticle initiators, with the goal of preventing migration and as an innovative method of photo-polymerization. The use of a nano-initiator also produces very well-dispersed hybrid materials. Depending on their bandgap, semiconductors such as ZnO have the potential to absorb UV-light-forming excitons (electron–hole-pairs) (Scheme 1).
 | ||
| Scheme 1 Summary of relevant schematic reactions for initialization by the nanoparticulate semiconductor ZnO and by commercial initiators. Weak bonds where fragmentation will occur are indicated by a dotted line. | ||
The radicals necessary for radical polymerization may be produced via the Photo–Kolbe reaction (reaction of a semiconductor hole with carboxylic acid, followed by decarboxylation), which was first reported by Kraeutler (1978).17 Kraeutler was also the first to demonstrate the initiation of methyl methacrylate polymerization in solution by alkyl radicals upon the decarboxylation of acetic acid by a semiconductor (TiO2-powder).18 For a long time, no results were published concerning any advantages of semiconductor-based polymerization in bulk systems. Initialization by ZnO nanoparticles in an isopropanol dispersion containing methyl methacrylate was investigated by Hoffmann et al. (1992)19 and Mills et al. (1994).20 At that point, polymerization rates seem to be far from any possible application (polymerization occurred after 90 min of irradiation). Similar studies have been published using other semiconductors, often without an inhibitor.21–24 The decarboxylation mechanism was first analyzed in 2010,25 and surface-modified ZnO nanoparticles for the initialization of bulk polymerization of acrylic esters were developed. For a non-optimized system, the reactivity relative to the weight content of initiators in the reaction mixture is only approximately 10 to 50 times smaller than that of molecular fragmenting initiators. These nanoparticulate initiators, based on ZnO modified with carboxylic acids, react via a fragmentation mechanism similar to that of common molecular initiators such as 1-[4-(2-hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propane-1-one (Irgacure®2959) (Scheme 1). This type of initiator is called a fragmenting or “Norrish type I” initiator. For modified ZnO-based systems, the effects of the stabilization of the fragmentation product R˙ and the photo-activity were first investigated in a previous study.26 Initiators that are characterized by non-fragmentation, such as the well-known initiators benzophenone or isopropyl-thioxanthone (ITX), are called “Norrish type II” initiators (Scheme 1). A combination based on a ketone, e.g., benzophenone, and a co-initiator, e.g., an amine, often MeEt2N, are necessary for these systems. During irradiation, the interaction between the ketone and co-initiator produces an aminoalkyl radical (initiating radical) and a ketyl radical through an electron/proton transfer mechanism (Scheme 1). One focus of the present study is the development of a novel material, based on ZnO nanoparticles, that does not fragment but produces an easily measured effect without the addition of any co-initiator. The recently developed “Norrish type I” ZnO modified with levulinic acid is used as a nanoparticulate reference. This initiation system also lacks any aromatic functionality. As mentioned above and shown in Scheme 1, the modified ZnO leads to a Photo–Kolbe reaction, as demonstrated in previous studies.26,27 The remaining electron, the holes and the trapped polymeric radicals are detectable by ESR28,29 (Fig. S12†), and for semiconductors, possible signals have been reported in previous studies.28,30–33 Hence, the theoretical motivation leading to the novel research on non-fragmenting nano-initiators presented herein was born by synthesizing an electron trap from an additional compound, such as a metal or metal ion (Scheme 1). Enhancing the lifetime of the hole should reduce the recombination of the exciton and therefore enhance the effectiveness of polymer initiation. Moreover, the systematic evaluation of the reactivity of doped particles without surface modification leads to a novel system with significant initiation potential, without a co-initiator or fragmentation. Additionally, this publication provides advanced and full analyses of the systems/curing and a detailed description of the synthesis procedure that produces easily surface-modified nano-ZnO, whose size is affected by the chosen solvent.
Materials and methods
Preparation of n-ZnO
The nanoparticulate ZnO used for the systematic studies described herein was formed by an injection procedure at room temperature using inexpensive and easy-to-handle precursors (Table 1).25 First, 0.15 mol ZnCl2 (purity ≥98%, Sigma-Aldrich, Germany) and 0.3 mol NaOH (Sigma-Aldrich, Germany) were dissolved separately in 180 mL methanol (with a cooling period required for the NaOH solution). Precipitation by injection of the NaOH solution into the ZnCl2 solution was performed at room temperature with fast stirring. Modifications of the ZnO with Cu(II), Pt(II), Mn(II) and Fe(III) were achieved by co-precipitation. The mixture was stirred for a minimum of 12 hours at room temperature. It was then centrifuged for 10 min at 4500 rcf (relative centrifugal force), and after the decanting of the solvent, the formation of two solid layers was clearly observed (Fig. S1†). The upper layer contained mainly the ZnO particles and the lower one mainly NaCl (confirmed by XRD analysis). The upper layer was separated and washed twice by dispersion in methanol or ethanol, centrifugation, and decantation to remove the remaining NaCl. Excess alcohol was removed by decanting after a final centrifugation (10 min, 4500 rcf), resulting in a white (in the absence of metal salts) paste of nano-crystalline ZnO without surface modification. After the precipitation either a (surface) modification or the dispersing in the resin were performed. For modification, the ZnO paste was dispersed in 100 mL of ethanol and heated to 80 °C under reflux. Subsequently, the surface modifier (31 mmol in 10 mL ethanol) was added to the reaction mixture, which was maintained at 80 °C for 1 h. The reaction mixture was then cooled under magnetic stirring and centrifuged (10 min, 4500 rcf). The solvent was decanted off, and the solid was washed twice with ethanol (which involved dispersion, centrifugation, and decantation).| Modification | Quantity for synthesis [mol%] | Color of the paste | Solid content [%] | Curing by lab.-unitc | Crystallite size (ZnO) [nm] | Washing remarks |
|---|---|---|---|---|---|---|
| a Loss of solid during synthesis (dispersion or solution). b 1 mol% NaOH added during the modification. c Manganese and iron produce only a moderate to small effect, as measured using a Hg mid-pressure system. Note that for the laboratory curing unit and the test printing applicator, only approximations are given; a commercial molecular “Norrish-type II” initiator would be classified at least by “++++++”. d Film fabricated by a 0.04 mm wire-bar applicator. e The Fe(III)Cl3 samples were only washed twice with EtOH; therefore, the XRD diffraction patterns clearly show significant amounts of NaCl. | ||||||
| Pt(II)(NO3)2 | 0.7 | Yellow-orange | 25 | +++ | 9.7 ± 1.7 | MeOH |
| Pt(II)(NO3)2 → Pt(0) | 0.7 | Brown-grey | 15 | ++ | 11.3 ± 1.4 | MeOH |
| Cu(II)Cl2 | 1.0 | Blue | 5.2 | + | 5.8 ± 0.9 | MeOH |
| Levulinic acida | 17.0 | White | 24 | +++ | 7.1 ± 1.1 | MeOH |
| Clearing up | ||||||
| Levulinic acida | 4.4 | White | 56 | +++ | 6.1 ± 0.9 | EtOH |
| Levulinic acida | 8.5 | White | — | +++ | 7.0 ± 0.6 | EtOH |
| NaOHb | ||||||
| Mn(II)Cl2 | 0.5 | Brown | 5.7 | n.m. | 8.7 ± 0.5 | MeOH |
| Mn(II)Cl2 | 1.6 | Brown | 5.4 | 0 (+) | 7.7 ± 0.6 | MeOH |
| Mn(II)Cl2 | 2.1 | Brown | 5.2 | n.m. | 7.7 ± 0.5 | MeOH |
| Cu(II)Cl2 | 2.0 | Blue | 5.7 | 0 | 8.5 ± 0.7 | MeOH |
| Fe(III)Cl3 | 0.24 | Brown | 28.3 | [0]d | 9.0 ± 1.3 | NaCle |
| Fe(III)Cl3 | 0.49 | Brown | 29.1 | [0]d | 8.1 ± 1.5 | NaCle |
| Fe(III)Cl3 | 0.74 | Brown | 12.6 | [0]d | 8.7 ± 1.3 | NaCle |
| Fe(III)Cl3 | 0.99 | Brown | 23.0 | [0]d | 9.1 ± 1.7 | NaCle |
| Fe(III)Cl3 | 1.24 | Brown | 21.0 | 0 | 8.5 ± 0.7 | NaCle |
Modification with Pt(0) was performed by an additional irradiation procedure. In this case, the freshly prepared n-ZnO was dispersed in 450 mL ethanol with 1 mmol Pt(NO3)2 and irradiated for half an hour in a UV-RS-1 reactor system (Heraeus Noblelight, Hanau, Germany), equipped with a TQ 150 UV immersion lamp (input rating 150 W). A color change from yellow-orange to brown-grey marked the reduction of platinum(II) to platinum(0). Before dispersion in the monomeric acrylic ester, the solid was separated and washed twice with ethanol, as explained above. Early results concerning variations, such as those of in situ monitoring and flow synthesis, are described in the Discussion section. All chemicals and solvents not specifically mentioned were of standard quality.
Characterization of n-ZnO
In addition to the in situ curing experiments (see below), characterizations such as TEM, IR, UV-vis (ex situ and in situ), curing of printed samples by laboratory units (IST Metz, Germany) and XRD measurements were performed. For the ex situ curing experiments, which will not be explained in detail, printed films (0.001 mm) and films fabricated by a wire-bar applicator (0.01 mm) were irradiated step-by-step by laboratory unit, and tacticity tests were performed. For XRD analysis, the full width at half maximum (FWHM) was determined using pseudo-Voigt functions, similar to eqn (2) through (4), using up to seven reflexes (Fig. 3), and the well-known Scherrer equation was used to calculate the crystallite size (with the shape factor defined as being equal to 1.0). The aspect ratio can be estimated from the crystallite size, which is calculated from the (100) and (002) diffraction peaks,34 if no significant amount of NaCl is measured.Procedure for the curing experiments
Immediately after fabrication, the methanol- or ethanol-containing initiator paste was mechanically dispersed (Ultraturrax) in a mixture of 75 wt% epoxy acrylate oligomer (3R1532, provided without pigments by Zeller&Gmelin GmbH&Co, Eislingen (Germany), used as received) and 25 wt% di-trimethylolpropan-tertraacrylate (DTMPTA). The additional solvent was evaporated under reduced pressure, resulting in the calculated wt% quantity within the resin in Table 2. The resin 3R1532 is an acryl-bisphenol-A-glycerolate modified with a fatty acid of moderate polarity. Additional information about the resin in comparison with other typical resins has been published elsewhere.29In situ monitoring of the irradiation-dependent double bond content was achieved by Raman35 and by Raman UV-vis spectroscopy36 (Fig. S4†). Note that the recently introduced automatic Raman UV-vis system has the unique ability to monitor both the double bond content (deep curing by Raman, Fig. S4†) and solidification (area curing by UV-vis, Fig. S5†). Due to the construction of the curing cells, a 310 nm high-pass filter (Duran glass) was used for the Raman spectroscopy measurements, whereas no filter (fused silica) was used for the Raman UV-vis measurements. All samples were sufficiently transparent to avoid heating by the 1064 nm Laser (1 W) of the Multiram FT-Raman spectrometer (Bruker Optic, Eislingen, Germany). UV-vis detection was performed using a MultiSpec Desktop/USB system (tec5 AG, Oberursel, Germany). For illumination, a specially designed flat cell (thickness 0.050 mm, glass for Raman and fused silica for Raman UV-vis; Fig. 2 in the ref. 26) was coupled via an optical fiber (0.4 mm diameter) to a Perkin Elmer optoelectronics xenon flash light (LS-FX) FX 1160 (maximum output 0.5 J per flash, regular 375 mJ per flash, 66.6 Hz). Absolute and spectral calibrations were performed using an absolute and spectral calibrated fiber optical receiver, according to the procedure recently described.37 The approach is summarized as follows. The receiver was calibrated relative to the solar spectrum at 06/07/13, 09:06 AM (true local time). The time was determined by a statistical procedure,37 and a power of 821.3 W m−2 was measured by a nearby meteorological station. Comparing this spectrum with the scaled ASTM38 spectrum (Fig. S3†) results in a detector-specific correction function A(λ), | (1) |
| Modification | Quantity within the resin [wt%] | Ramana | Raman | UV-visb | M ∞ [%] | n total [103 flashes] | |||
|---|---|---|---|---|---|---|---|---|---|
|
t
i
:tf [103 flashes] |
M ∞ [%] | n total [103 flashes] |
t
i:tf [103 flashes] |
t
i:tf [103 flashes] |
|||||
| a A glass filter (high-pass, 310 nm) was used in the measurements. The manual measurements resulted in large deviations (Fig. S4), which are indicated by > and <. b Raman and UV-vis monitoring was performed during the same experiment with the same sample.26 Only the reference and the acid-containing systems were freshly dispersed/prepared. The metal-containing nanoparticulate systems were stored in the acrylic mixture for approximately one year between the Raman and the Raman UV-vis measurements. c Total number of flashes used for the experiment. This number can be converted to irradiance (J m−2) (as discussed in the Materials and methods section, Fig. 5 and Fig. S3). d The final double bond content is undetermined (Fig. 4). | |||||||||
| Norrish type I | Levulinic acid 8.5 mol% | — | n.m. | n.m. | n.m. | 11.8:29.4 |
6.5:45.3 |
55 | 78 |
| 9.1:33.9 |
n.s. | 75d | 39 | ||||||
| 0.8 mol% | 5.7 | n.m. | n.m. | n.m. | 7.7:20.5 |
11.0:39.8 |
72 | 49 | |
| 4.4 mol% | 12.3 | n.m. | n.m. | n.m. | 10.4:37.2 |
36.1:82.1 |
68 | 78 | |
| 12.6:24.6 |
n.s. | 70 | 39 | ||||||
| Irgacure® 2959 | 2.0 | 32:40 |
69 | 49 | 3.4:14.8 |
1.9:4.7 |
51 | 49 | |
| Norrish type II | Pt(II)(NO3)2 | 3.6 | >10:<15 |
70 | 24 | 11.0:30.2 |
15.3:36.5 |
69 | 39 |
| Pt(0)a | 3.6 | >10:<20 |
70 | 42 | 42.7:89.5 |
41.2:102 |
73 | 98 | |
| Cu(II)Cl2 | 5.2 | 37:91 |
63 | 150 | 31.5:81.5 |
49.2:88.4 |
67 | 98 | |
| Mn(II)Cl2 1.6 mol% | 5.4 | >20:<60 |
62 | 60 | n.s. | n.s. | 100 | 78 | |
| Benzophenone | 2.0 | n.m. | n.m. | n.m. | n.s. | n.s. | 100 | 78 | |
Data evaluation of in situ curing experiments
The data obtained by the automatic in situ Raman measurements are fitted by multiple pseudo-Voigt functions PV(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ),
),| PV() = area (ηg() + (1 − η) l()) | (2) |
 | (3) |
 | (4) |
| f(t) = b − a exp(−exp(−k(t − ts))) | (5) |
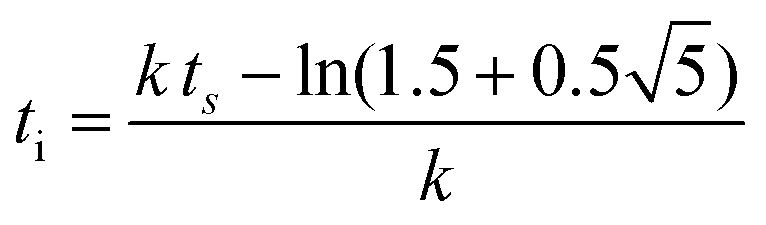 | (6) |
 | (7) |
Results
Syntheses/characterization and ex situ curing
The goal of the author was to perform a fast, high-concentration synthesis with easily accessible low-cost chemicals to allow for a possible scale-up for industrial application. The synthesis of the unmodified ZnO samples and the results of the ex situ curing experiments are summarized in Table 1. The first step is to perform the synthesis without any surface modifier and to prevent strong adsorbance onto the surface of the particles. Therefore, only chlorides and NaOH are used as starting materials (Table 1). Note, in particular, that the often-cited premise that organic acid salts can be used as precursors without foreign stabilizers is unsubstantiated. Researchers have seemed to ignore the fact that their model systems are flawed; the organic acid will interact with the surface, especially that of ZnO, due to its basic character (point of zero charge is approximately 9.4;39Fig. 6). This basic character of ZnO is also the reason for the very high stability of the organic acid's surface functionalities and their strong dispersion properties, which further depend on the dispersion medium. The stability of the modification is also analyzed by IR spectroscopy (Fig. 1). | ||
| Fig. 1 FTIR spectra of ZnO:levulinic acid (8.5 mol%) absorbed onto a ZnSe HATR-prism. The modifier40 (Table S1†), is clearly recognized after application and evaporation of the solvent, M1. Rinsing with ethanol and evaporation, M2, has no effect. Successive rinsing with NaOH (5 × 10−3 M) in ethanol and evaporation, M3, desorbs the particles from the ZnSe surface. Successive rinsing with HCl (5 × 10−3 M) in ethanol and evaporation desorbs/dissolves the whole system. Signals from residual ethanol (1050 cm−1) are visible for those nanoparticles without surface modification. Rinsing with aqueous solutions clearly affects the OH vibrational bands (>3000 cm−1). | ||
A paste containing ZnO is placed on the ZnSe HATR-prism (size 20 mm × 55 mm). After evaporating the alcohol, the measurement is performed. These few milligrams of adsorbed solid are rinsed several times with an excess of different solvents (>1 mL), eventually removing the modifier from the ZnO surface (Fig. 1, top). If using organic acids in combination with the ZnO, the changes in the spectra are explained by desorption or solvation of the ZnO system from the ZnSe surface. Adsorption of the unmodified ZnO paste appears to be, in general, reduced, and the C–O stretching vibrational band (1050 cm−1) of the alcohol is clearly recognizable (Fig. 1, bottom). Successive rinsing results in a stronger adsorption of the sample onto the ZnSe and in modification of the particles with the organic impurities of the rinsing solvents (from the leaching of the wash bottle or the transfer pipettes). The adsorbed alcohol allows for the dispersion of the unmodified ZnO-containing paste after the final decantation. The suspension of the ZnO by organic acids also affects the synthesis (Table 1). For a modification with 17 mol% levulinic acid, the solution is observed to clear upon addition of the modifier, which most likely signifies additional solvation of the ZnO and not merely dispersion of the nanoparticles. As demonstrated by TEM images (Fig. 2 and Fig. S22†), levulinic acid can produce perfectly dispersed nanoparticles in ethanol.
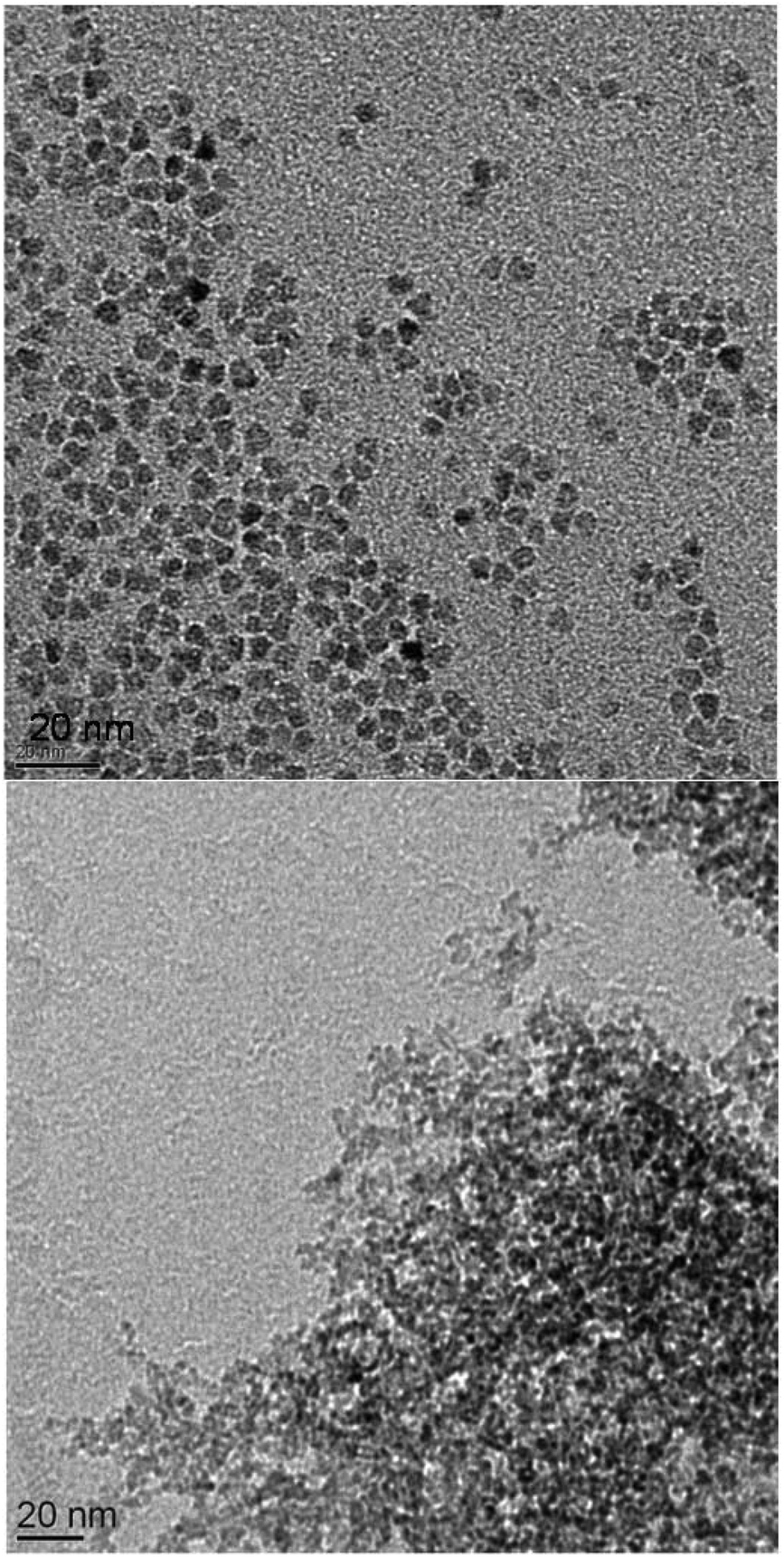 | ||
| Fig. 2 TEM image of ZnO:levulinic acid (8.5 mol%) on the right and of the ZnO:Cu(II) (1.0 mol%) on the left. Additional images are presented in the ESI, Fig. S15–S23.† The size distribution of the spherical ZnO:levulinic acid (8.5 mol%) particles is determined via counting and statistical analysis, and the average diameter of the particles is determined to be 5.1 nm ± 1.0 nm (Fig. S14†). | ||
Both the content of the levulinic acid used and the complexity of this apparently simple synthesis (see discussion) produce variations in the composition of the solid (Table 1) and in TEM images (Fig. 2). XRD analyses (Fig. 3) were performed to verify the crystallinity and determine the crystallite size (Table 1). All presented precipitation syntheses (Table 1) produced crystalline particles, which are important for the absorption of irradiative light and reactivity, smaller than 10 nm of only slightly different sizes (Fig. S2†). Additionally, no significant differences in the FWHMs between the different directions (200 and 100) were observed for the samples without NaCl. Hence, spherical particles with aspect ratios of approximately 1.0 ± 0.3 radii were prepared.‡ Co-precipitation in the presence of metal salts appears to affect the solid content of the final paste, but the effect is weaker than that observed when using a levulinic acid as the modifier. Insertion of specific foreign atoms into the crystal lattice cannot be conclusively demonstrated, even after sintering the ZnO:Cu(II) (1 mol%) particles.
 | ||
| Fig. 3 XRD diffraction pattern of nanoparticulate ZnO:Cu(II) (1 mol%). Fitting by pseudo-Voigt functions results in the FWHMs. A superposition of the first three reflexes is a clear indication of very small-grain crystalline nano-ZnO, also seen in Fig. S2.† | ||
In situ UV-vis measurements (d = 0.2 mm) were performed to investigate the time needed for completion of the reaction. The reaction could be monitored by UV-vis for approximately 3.5 s, until the clouding was too pronounced (Fig. S6†). At this time, the intended product bandgap of approximately 350 nm was nearly reached (Fig. S7†). This experimental finding is an interesting result that will be addressed in the discussion. Ex situ curing experiments were performed via a tacticity test, as explained in the Materials and methods section (Table 1). The coloration of the pastes is in general not recognizable for the 0.002 mm (printing), for the 0.05 mm (in situ monitoring) and also for the 0.5 mm thick layers (ESR spectroscopy), Fig. S8–S10.† Additional testing on an industrial printing machine showed that colored resins (color scale CMYB) containing up to 50 wt% benzoyl benzoic acid-modified ZnO paste could be printing without affecting the image quality.
In situ curing experiments
Only those samples that led to an effect with the laboratory curing unit (Table 1) were analyzed by Raman and Raman UV-vis (Table 2 and Fig. 4). Additionally, different attributes of this resin mixture were verified:1. No polymerization without initiator (<150k flashes)
2. No polymerization with 4 wt% unmodified nanoparticulate ZnO (<90k flashes)
3. No signal change, minimal macroscopic curing with 2 wt% benzophenone (<70k flashes)
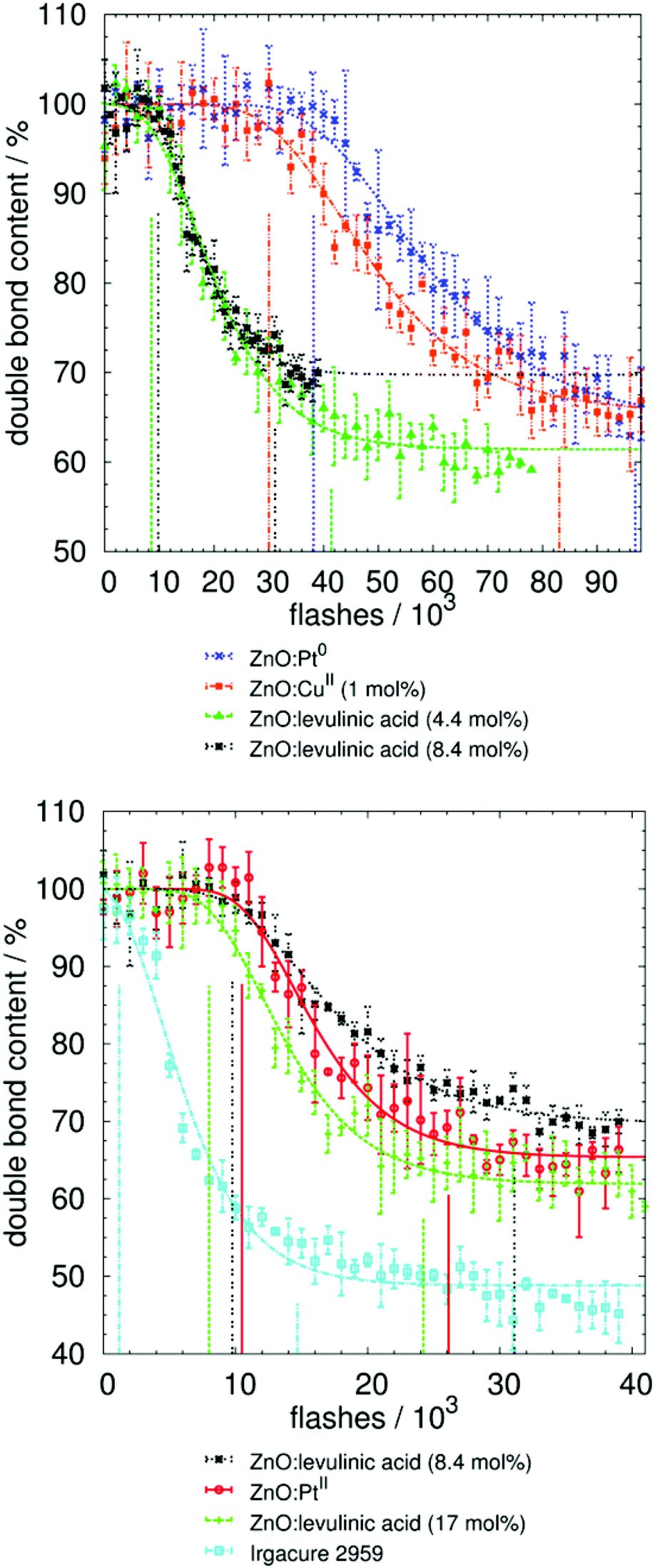 | ||
| Fig. 4 Results and fitting of photo-polymerization measurements by Raman UV-vis spectroscopy. Initial and final times (in number of flashes) are marked by vertical lines. This number can be converted to irradiance (J m−2), as discussed in the Materials and methods section (Fig. 5 and Fig. S3†). Neither the un-modified ZnO nor the non-fragmenting benzophenone resulted in curing, whereas the ZnO-containing copper ions and, especially, the ZnO-containing platinum ions resulted in curing rates nearly as fast as the ZnO modified with levulinic acid. The molecular fragmenting initiator was approximately 10 times faster, taking into account the lower weight content of initiator in the reaction mixture. | ||
Consequently, it was possible to measure the effects and differences in the curing potential for all investigated initiators (Tables 1 and 2). The stability over time was also assessed, as a result of the time delay between the Raman and the Raman UV-vis measurements.26 The two monitoring systems did not operate simultaneously because components of the Raman system were used in construction of the Raman UV-vis system. As a result, a storage time of approximately one year between the Raman and Raman UV-vis measurements was imposed for all metal-containing samples. This result demonstrates that the acrylic mixtures for which reactivity is proven by both systems are stable for at least one year (Table 2).
Discussion
Synthesis and characterization
The most important aspect of this work is the introduction of a possible application for nanoparticulate semiconductors. Thus, a focus of this work was the demonstration of curing, which is discussed in greater detail in the next section. To aid the reader in approaching this topic as a new research avenue, the syntheses and some modifications thereof are discussed in this report. The one- and two-step syntheses performed in this work are simple (co-precipitation of precursors and modification) and result in a large amount (0.4 to 0.5 mol L−1) of similar or comparable samples for testing the photo-reactivity. The addition of surface-active compounds during ZnO formation can be useful in further optimizing the yield or properties of the system; however, for reasons of comparability, such addition was not performed in the present study. For example, using surface-active modifiers during precipitation can lead to variations in morphology.41 The ability to produce perfectly dispersed nanoparticles, (Fig. 2, Fig. S14† (statistical analysis) and S22† (TEM) using this one- or two-step synthesis proves that, under the appropriate experimental conditions (aspects related to timing and experience) the aggregation of non-modified ZnO is reversible. Two important factors that appear to affect the final reactivity have to be considered:• crystallite size
○ direct size effect = number disadvantage
○ indirect size effect = bandgap effects (absorbance blue shift)
• dispersion quality = surface effect
The so-called “direct size effect” or “number disadvantage” is an unfortunate side effect of the increased mass of the initiating species. One sphere of ZnO with d = 5 nm is 700 times heavier than one molecular initiator (300 g mol−1). This disadvantage increases with d3 such that a diameter of 8 nm, for example, is heavier by a factor of 3000. This relationship explains why the initiation potential of nanoparticulate semiconductors for bulk polymerization remained undiscovered for so long, as mentioned in the introduction. The size-induced bandgap shift19,42 is not significant when using a xenon light. This observation may be due to the broad wavelength distribution of the irradiation source used in this study36 (Fig. 5). For industrial processes, mainly mid-pressure Hg lamps are used, which have specific and very discrete irradiation signals. In this case, the bandgap shift43 must be closely monitored because the absorption limit of n-ZnO occurs at approximately 350 nm (Fig. S7†) and is therefore attuned to the strong 365 nm signal of mid-pressure Hg lamps.
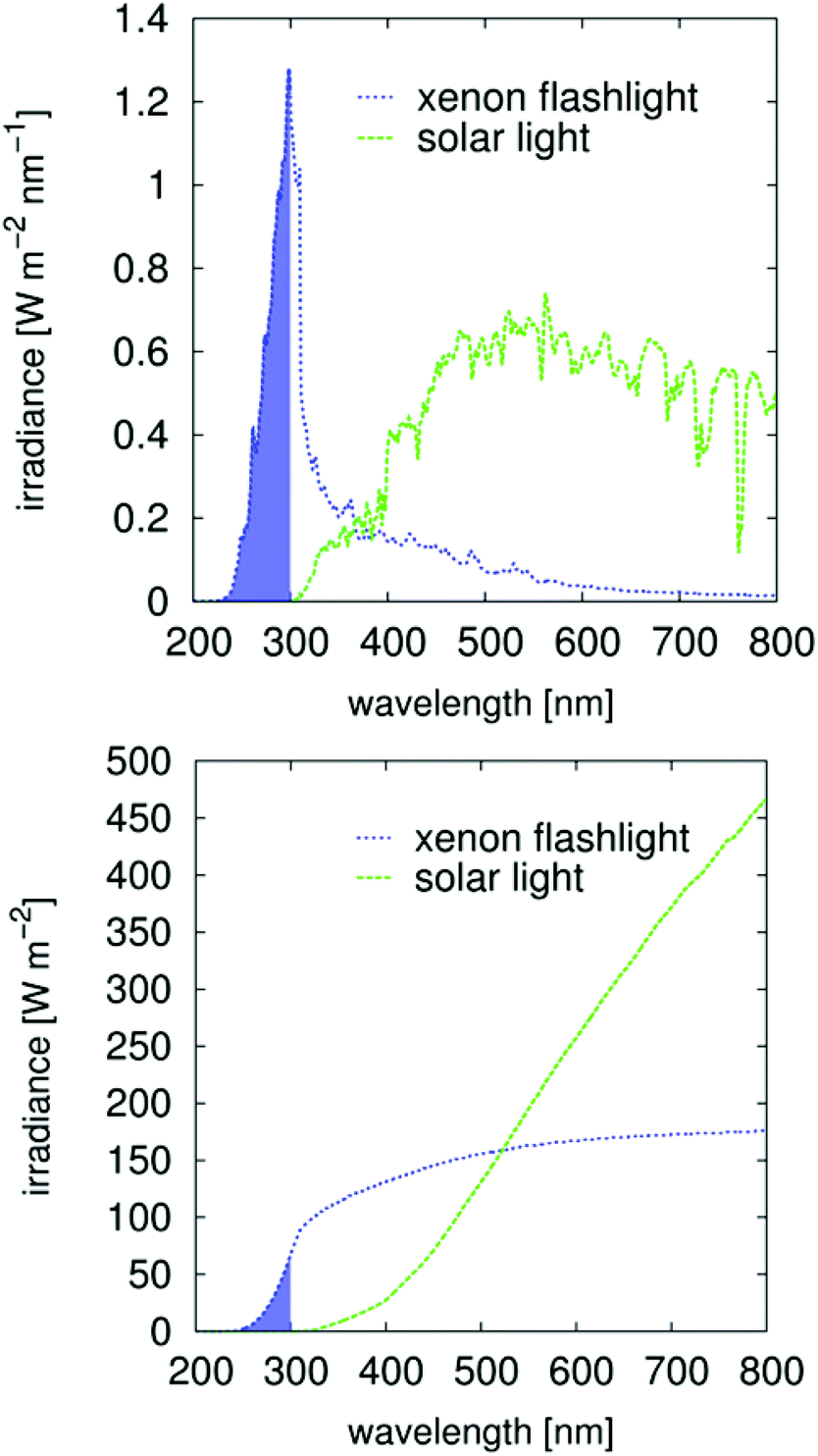 | ||
| Fig. 5 Results of the absolute and spectral calibration of the fiber optical flash light. The filled range is influenced by the absorbance of the resin, the absorbance of the glass filter and is beyond the calibration range of the fiber optical receiver. | ||
The solvation of the starting material, of the precursors and the charge stabilization/dispersion of the final particles (Scheme 2), are major factors, the effects of which are beyond the scope of this paper. The semiconductors fabricated by the procedure (Table 1) were mostly well-dispersed, resulting in transparent, colorless, cured, acrylic mixture films, even for those with thicknesses of up to 0.5 mm (Fig. S8–S10†), which therefore resulted in well-dispersed colorless hybrid materials. Note that these cured samples after a high-dose irradiation with a 500 W continuous xenon flash light results in no recognizable coloration, Fig. S8–S10.† Effects on the synthesis that most likely derived from different additional parameters, such as the water content and the purity of the chemicals, are difficult to identify. Additionally, the crystallite size can be affected by the solvent during precipitation. Pure, unmodified spherical ZnO with diameters as small as 4.1 nm can be synthesized using methanol as a solvent, whereas ethanol and isopropanol generally result in larger spherical crystallite sizes of approximately 10 nm. These effects on the formation kinetics and the dispersion stability are also correlated with the solubility of NaCl and NaOH. Cleaning by washing with pure solvents only results in the complete removal of NaCl for MeOH (Table 1). The effects of NaCl on the formation kinetics seem to be significant. Influences of water on the formation of the neutral precursor (ZnOH2) most likely exist. For example such effects of water (as impurity or from the atmosphere) are clearly proven in a spectroscopic study for the siloxane formation.44 Following to Scheme 2 in the case of the ZnO formation the formed water can lead to an autocatalytic reaction which explains the possibility to suppress the ZnO formation/precipitation if smaller concentrations of starting material are used. The reaction is completed in less than 5 s, which is interpreted as the time required to mix the reactants (Fig. S6 and S7†). Note that calcination is not necessary to obtain crystalline particles. The spectroscopic studies of Bahnemann et al. (1987),42 performed using Zn acetate as a precursor, indicated much longer reaction times: 20 min for a product bandgap of 355 nm or 2 h for a product bandgap of 365 nm at 65 °C. Direct comparison is not possible because the abovementioned authors used a smaller concentration of only 1 mmol L−1 Zn(OAc)2 and a substoichiometric concentration of NaOH, but it can be concluded that the formation of NaCl is a driving force of the reaction. Due to solubility effects, Ostwald ripening appears to be suppressed, resulting in small, uniform particles with a bandgap of approximately 350 nm (Fig. S6 and S7†). After precipitation, the reaction mixture can even be stirred overnight without further growth of the particles. Precipitation in water results in larger, non-phase-pure ZnO particles. A substoichiometric concentration of NaOH, as used in the abovementioned reference,42 results in positively charged nanoparticles, which are modified by acetic carboxylate ions present in solution. The unmodified ZnO is even able to adsorb ethanol and extract organic impurities from the rinsing solvents used in the IR experiments (Fig. 1, bottom). An adsorption study by Degen and Kosec45 confirmed that positive surface charge is important for the adsorption of modifiers onto powdered material.
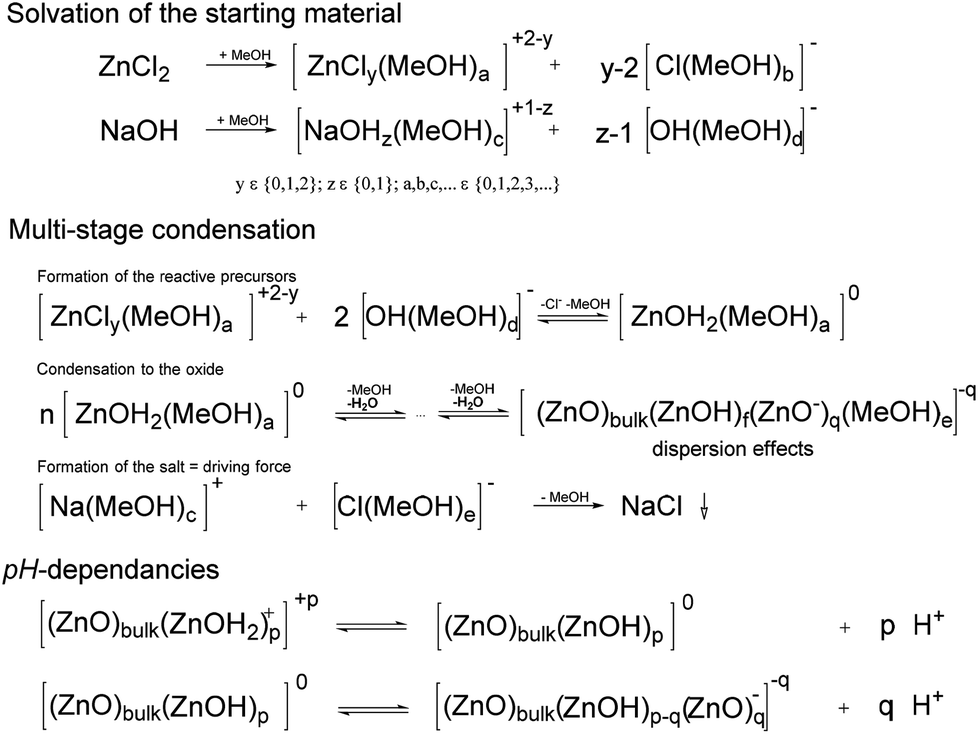 | ||
| Scheme 2 Schematic for solvation and condensation during ZnO formation. The driving force of the reaction is precipitation of the “non-soluble” NaCl. The protonation/deprotonation of the surface are also schematically shown. | ||
The proton activity and the activities of the protonated and deprotonated modifier in non-aqueous systems are not measureable. In aqueous systems of unmodified ZnO, zeta potential measurements were performed, which can be compared with the calculated relative charge (i.e., the relation between the content of the acid and of the carboxylic ion) (Fig. 6). These measurements are in accord with the known pH dependencies of the ZnO particles and the possibility of charge reversal. It can be assumed that, during the condensation presented in this study, the particles are neutral or slightly negatively charged (Scheme 2). Protonation during (due to) surface modification results in a reversal of the particle charge, followed by very strong adhesion of the modifier onto the particle surface (Fig. 1). A reduction in the crystallite size, due to the modification, is also likely for ZnO. The increase in viscosity during the mixing of the modifier solution into the dispersion containing the unmodified ZnO is an additional indication of charge reversal. On the one hand, for the non-surface modified ZnO, free access to surface hydroxyl groups, indicated in IR measurements by the strong OH vibrational bands at approximately 3330 cm−1 (Fig. 1), is likely necessary for reactivity and dispersion stability. On the other hand, hydrogen bridges between the OH functionalities of the resin and the surface of the semiconductor (e.g., ZnO−) will definitely affect the dispersion stability, as well as the possibility of producing polymerization-inducing radicals (Scheme 1). The literature also confirms the effect of hydroxyl groups on photo-reactivity. For the oxidation of isopropanol with TiO2, Kobayakawa et al. (1990),48 came to the conclusion that the photocatalytic activity increases with the concentration of surface hydroxyl groups before decreasing from an inflection point. An inverse dependence of the oxygen produced by TiO2 dispersed in water on the surface concentration of hydroxyl groups was observed by Oosawa and Grätzel.49,50 For the presented non-fragmenting system in particular, the surface charge will most likely affect the reactivity of the initiator. In the following, possible variations of the synthesis, which may ultimately have the potential to enhance reactivity and reproducibility, are summarized. As mentioned in the Materials and methods section, the manual removal of the NaCl-rich phase (Fig. S1†) and washing with methanol results in phase-pure ZnO. Likewise, additional washing of the surface-modified samples with ethanol will eliminate NaCl, whereas the doped particles which were washed only one cycle contain NaCl (Table 1). Water, as impurities in the solvents or formed (Scheme 2), definitely affects the solubilities. Newer studies involving a single washing of the unmodified paste using 1:1 mixtures of water and ethanol indicate that removal of NaCl is possible without using methanol, but potential effects on aggregation have yet to be evaluated. Optimization of the modifiers, content and preparation conditions remains to be accomplished for the system to be fully understood and controlled. Other factors to be considered are the purity (as mentioned above) and the stability of the chemicals. For example, ZnCl2 can form ZnO, and NaOH absorbs both water and carbon dioxide, which can result in a slightly green solution in ethanol. A promising approach involves separating the NaCl before ZnO formation. It should be possible to first remove the NaCl after the reaction of the ZnCl2 solution with an alcoholate such as sodium isopropanolate. The water-free solution of zinc isopropanolate will react with water to form the ZnO. Due to the lack of proper equipment for keeping the first step of the synthesis water-free, only a small fraction of crystalline ZnO (13 nm) was separable by the first test synthesis. To explore the possibility of scaling the synthesis up, simple flow syntheses were also performed. The fast precipitation (<5 s) in ethanol (Fig. S6†) led to larger particles with crystallite sizes of 19.7 nm ± 3.3 nm, which were less reactive relative to the same weight content of initiators in the reaction mixture produced by the unvaried synthesis (see above). The precipitation resulted in an improvement in viscosity, followed by blockages in the flow precipitation reactor. This problem should be solvable, e.g., by applying jet systems.
 | ||
| Fig. 6 Zeta potential measurement of unmodified colloidal ZnO nanoparticles in water, calculated relative charge of acetic acid (in water) and isoelectric points reported by select references (arrows; ref. A;46 ref. B;47 ref. C39). Within the hashed pH range, the nano-ZnO is positively charged, whereas a significant amount of the acid is negatively charged. α-Oxo carboxylic acids are stronger acids than acetic acid (due to their resonance stabilization). | ||
To summarize, highly reproducible batch syntheses producing large amounts (>0.4 mol L−1) of alcohol-stabilized or short-chain-acid-modified reactive crystalline nanoparticles for transparent and clear (colorless) dispersion in polar solvents (specifically, acrylic ester) are an interesting finding of the presented study. Optimization of the synthesis can also result in perfectly dispersed nanoparticles (Fig. 2, Fig. S14 and S22†).
In situ curing experiments
Most likely due to the “direct size effect” discussed above, the initiation potential for the bulk polymerization of nanoparticulate fragmenting ZnO systems was reported as recently as 2012.25,26 Non-fragmenting nanoparticulate initiators, first presented in the current paper, are obtained by combining the nanocrystalline ZnO with small amounts of integrated or attached compounds. Thus, to prevent exciton recombination of the semiconductor (Scheme 1), small quantities of metal ions (Pt(II), Cu(II)) or metal (Pt(0)) are combined with nano-ZnO, which should lead to electron trapping. Because the resin containing unmodified nanoparticulate ZnO undergoes no curing under a xenon flash light over fewer than 90000 flashes, all of the varied ZnO particles (fragmenting and non-fragmenting) result in an enhanced initiation potential (Fig. 4 and Table 2). Without a high-pass filter, the potential of the particle-based initiators for either deep curing (Raman) or area curing (UV-vis36) is not as high as that of the initiators available from Irgacure®2959. The final monomer content, M∞, is calculated from the parameters in eqn (5) and is always checked for the slope in the monomer content after curing (Fig. 4). The M∞ of this acrylic ester resin mixture, obtained via particle-based systems, is higher than the M∞ resulting from initiation by the molecular “Norrish type I” initiator (Table 2, Raman UV-vis). This result is in accord with the expected lower mobility of the smaller number of initialization compounds (due to the increase in mass by a factor of >3000), which also typically affects the ESR spectra10 (Fig. S11 and S12†), and can be interpreted as an additional immobilization of the nanoparticle-based initiator. For ZnO:Cu(II), a reduction in the typical ESR signal (Fig. S11†) due to electron transfer is also detectable. The reactivity of the holes is also experimentally manifested by photo-reactivity, as explained in detail in Schmitt et al., 2012.25 In this case, 1-undecanol reacts after a first electron transfer from an un-doped ZnO to form undecanal. Both species are measured and identified by GC-MS. Hence, it can be concluded that, for these ZnO:M systems (M = Pt(II)/Cu(II)), it is likely that the holes generated by the semiconductor are able to react with the hydroxyl groups of the monomers to form intrinsic initiating radicals (Scheme 1). This observation is in clear contrast to the behavior of non-fragmenting molecular initiators, such as the classical “Norrish type II” initiator benzophenone, which require a co-initiator. As expected, verification of the abovementioned effects, using the initiator benzophenone, results in hardly any curing under similar conditions. The Raman measurements presented in Table 2 are not quantitatively comparable with the Raman UV-vis measurements due to construction characteristics (Raman incorporates a high-pass filter composed of Duran glass), the time between the measurements and the geometry of the sample cell (differences in distribution of the irradiation). For the particle-based initiators, the effect of the high-pass filter is very low, whereas, for the molecular initiator, the effect is extreme (Table 1). Following to the supplier, the maximum absorbance of Irgacure®2959 occurs at approximately 280 nm, reaching up to 340 nm for 0.1% in acetonitrile. UV-vis transmission measurements of a saturated solution of Irgacure®2959 in cyclohexane (d = 10 mm) show no additional absorbance above 300 nm (due to insufficient miscibility). Interestingly, the “time” required for the reaction to start (i.e., the inhibition period) increases from 3.4 × 103 to 32 × 103 flashes (multiply by 1.0 J m−2 per flash to obtain the relevant irradiance), whereas the curing (ignoring differences in M∞) proceeds with similar doses of light (Table 2) (the resolution and area are difference for each measuring cell). Such differences are very interesting with respect to the optimization of molecular or particulate initiation systems, for example, the optimization of wavelength sensitivity. These differences are only simultaneously detectable in one experiment by the recently developed Raman UV-vis system. Again, the tested molecular initiator produces enhanced area curing with the higher energy light (Table 2) compared to the depth attained by Raman spectroscopy and the area curing of UV-vis spectroscopy. The higher energy light has the lower penetration depth due to absorption by the resin and the initiator. This finding and the similar M∞s imply that the ZnO systems (Table 2) primarily operate with light in the range of the bandgap (350 nm) not absorbed by the acrylic group (299.5 nm, absorbance >1),29 presenting a possible application of the material as an initiator under LED irradiation. First, measurements of specific irradiation wavelengths demonstrate that systems containing ZnO modified with benzoyl formic acid are thoroughly cured, even by irradiation across the range of 371 to 379 nm, at the very base of the bandgap. Absorbance by molecular initiators, especially those used in LED applications, occurs at wavelengths longer than 400 nm such that a yellowing of transparent films (lacquers) occurs. The sharp bandgaps characteristic of semiconductors are another typical, unique property of nanoparticle initiators that leads to multiple research opportunities which results in no yellowing or other coloration of the system. Even the photo degeneration of the ZnO seems to be suppressed which is known to occur in liquid water. In the future, the reactivity of the semiconductor ZnO can be altered by modifying the particle size51 and morphology52 and incorporating additional modifiers such as electron-trapping nitro compounds.53 Other effects that remain to be investigated are sorption properties54 and the relations between modification and the resin used. The dispersion associated with the interaction and radical transfer between the particle system and the curable monomers is a particularly interesting field of research. Additional reactions of the nanoparticles can also take place, such as acid-induced ester cleavage of the acrylic ester monomers, which must be closely monitored. The monomeric acrylic esters are widely varied in terms of functionalization and, therefore, in polarity. But acrylic esters are not the only photo polymerizable systems. Hence, the modifier and the doping can be adjusted for the resin, or the entire curing process (resin, conditions and hardware) could even be adjusted for the nanoparticle system.
Conclusion
The synthesis (>0.4 molar) of modified bi-functional crystalline ZnO nanoparticles is described in detail. The synthesis produces stable dispersions in polar solvents, such as polar acrylic ester resins, and the particles vary in reactivity via photo-initiation. Test prints of pigmented resin (CMYB) result in no interference of the printing image. Additionally, layers measuring at least 0.5 mm, cured to form transparent solids by irradiation with a 500 W continuous xenon light, are formed without recognizable yellowing or other coloration (Fig. S8 through Fig. S10†). First, variations in the synthesis with respect to solvents, the content of the modifier or preparation conditions (flow reactor) and the analysis of the effects of these changes on the products are presented within this study. Low-solubility compounds (NaCl, precursors and products) appear to suppress ZnO growth and to result in a fast reaction within the range of mixing times of the starting solutions. The investigated transparent model resin is commonly used in off-set printing processes. Due to their size, nearly all common molecular initiators result in migration problems, which is a major problem associated with UV-induced polymerization. The results of in situ analyses of curing clearly demonstrate the potential of the recently introduced Raman UV-vis system17 to simultaneously measure both deep curing and area curing in situ and to monitor the dependence of the penetration depth of illumination on polymerization. The UV-vis solidification is can even be applied for non-transparent systems. The present work also explained these novel analysis procedures including the absolute and spectral calibration of the light source in detail. The in situ curing experiments performed in this study show that the reactivity of the reported non-optimized, non-fragmenting n-ZnO:Pt(II) initiator is similar to that of the fragmenting n-ZnO initiator modified with levulinic acid, whereas ZnO fabricated by the same procedure without modification leads to no reaction. It has been shown in the publication that the first one is reactive due to the enhanced lifetime of the exition whereas the second one is reactive due to the attached reactive carboxylat ion. Both demonstrate similar photo-polymerization properties under UV-A illumination and UV-LED illumination, which require further investigation. However, even without the use of any co-initiators, most metal-containing systems demonstrate significant reactivity. Every known non-fragmenting initiator, e.g., benzophenone or ITX, requires a co-initiator. This point is emphasized because this difference is fundamental and of major importance as a unique attribute of potential nanoparticulate, non-migration “Norrish type II” initiators. Whether a co-initiator could further enhance the curing rate of one or both nanoparticulate systems remains an open question. An advantage of using ZnO is the fact that the nanoparticles, if consumed, will rapidly decompose in nearly any acid, resulting in non-toxic zinc ions and, eventually, platinum or other investigated compounds. Additionally, one nanoparticulate ZnO initiator with a diameter of 8 nm is more than 3000 times heavier than a single molecular initiator, whereas the difference in reactivity between the ZnO systems and the “Norrish type I” molecular initiator is in the range of 101. At the moment, the “Norrish type II” particle-based system, even without a co-initiator, appears to be too slow or too expensive in fabrication and development for commercial use. However, for particle-based systems, migration, particularly via diffusion or gas-phase transfer, is eliminated. With respect to human health, the possibility of a non-migration initiator composed of harmless compounds demands further investigation, for the sake of posterity.
Analyses of the attributes affecting the efficiency of the particles by varying the particle type, structure,34,41,52,55,56 surface modification,57 size51 and bandgap,41 or by changing the resin or process parameters, provide many avenues of further scientific and industrial research. The more acidic TiO2 (anatase) semiconductor, whose preparation, properties and photo-reactivity have been reported on in multiple studies,21,43,58–63 are interesting but unlikely to produce a stable system with tunable dispersion upon modification with organic acids (Fig. 6). The stable dispersion of the nanoparticles within the acrylic ester is a necessary condition for measurable initiation. It should therefore be possible to optimize the monomers, e.g., acrylic esters, to attain the properties of a perfectly prepared functionalized nanoparticle. Whether stability over time (e.g., more than one year in the resin) is an effect of the reactivity or of an energy barrier is another polymer physics topic that requires further investigation. Another interesting topic is the effect of the degradation of the potentially reactive ZnO (intended and unintended). Early results obtained from in situ ESR spectroscopy lead to the conclusion that the particles are integrated in a highly immobile,29 rigid matrix (Fig. S11 through Fig. S13†).
To conclude, the presented results and suggested possibilities present a wide array of topics for scientific research in nanoscience, nanotechnology and polymer science. It should be emphasized that optimized, modified or doped semiconductors can be both “Norrish type I” fragmenting and “Norrish type II” non-fragmenting initiators, with additional properties like corrosion and irradiation protection for the final hybrid-coating, green fluorescing which leads to additional applications.64–67
Abbreviations
| n.m. | Not measured |
| n.s. | No significant change in the signal |
Acknowledgements
Special thanks to S. Kuhn (experimental support), F. Heib and R. Hempelmann for their support.Notes and references
- G. Allegrone, I. Tamaro, S. Spinardi and G. Grosa, J. Chromatogr., A, 2008, 1214, 128–133 CrossRef CAS PubMed.
- S. Aprile, E. Del Grosso and G. Grosa, Xenobiotica, 2011, 41, 212–225 CrossRef CAS PubMed.
- H. Gallart-Ayala, O. Nunez, E. Moyano and M. T. Galceran, J. Chromatogr., A, 2011, 1218, 459–466 CrossRef CAS PubMed.
- M. Hefnawy, Advances in Food Protection Series A: Chemistry and Biology, Springer VCH, 2010 Search PubMed.
- T. Jung, T. J. Simat and W. Altkofer, Food Addit. Contam., Part A, 2010, 27, 1040–1049 CrossRef CAS PubMed.
- T. Jung, T. J. Simat, W. Altkofer and D. Fugel, Food Addit. Contam., Part A, 2013, 30, 1993–2016 CrossRef CAS PubMed.
- A. K. Malik, C. Blasco and Y. Pico, J. Chromatogr., A, 2010, 1217, 4018–4040 CrossRef CAS PubMed.
- T. Rothenbacher, M. Baumann and D. Fugel, Food Addit. Contam., 2007, 24, 438–444 CrossRef CAS PubMed.
- Bundesinstitut für Risikobewertung, Stellungnahme Nr. 044/2005 des BfR “Bestandteile von Druckfarben in Getränken aus Kartonverpackungen”, 2005 Search PubMed.
- Bundesinstitut für Risikobewertung, Stellungnahme 028/2008 des BfR “Ersatz von Isopropylthioxanthon (ITX) in Druckfarben durch nicht bewertete Stoffe ist nicht sachgerecht”, 2008 Search PubMed.
- J.-P. Fouassier, Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications, Hanser Gardner Pubns, 1995 Search PubMed.
- BASF, Safety Data Sheet Irgacure® 2959, 2011 Search PubMed.
- A. G. Rahn, EnergyCuring product guide, 2013 Search PubMed.
- BASF, Safety Data Sheet Irgacure® MBF, 2013 Search PubMed.
- L. Angiolini, D. Caretti, S. Rossetti, E. Salatelli and M. Scoponi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5879–5888 CrossRef CAS PubMed.
- S. Jauk and R. Liska, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 804–810 CrossRef CAS PubMed.
- B. Kraeutler, C. D. Jaeger and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4903–4905 CrossRef CAS.
- B. Kraeutler, H. Reiche, A. J. Bard and R. G. Hocker, J. Polym. Sci., Part C: Polym. Lett., 1979, 17, 535–538 CrossRef CAS PubMed.
- A. J. Hoffman, G. Mills, H. Yee and M. R. Hoffmann, J. Phys. Chem., 1992, 96, 5546–5552 CrossRef CAS.
- Z. Y. Huang, T. Barber, G. Mills and M. B. Morris, J. Phys. Chem., 1994, 98, 12746–12752 CrossRef CAS.
- C. Damm, J. Photochem. Photobiol., A, 2006, 181, 297–305 CrossRef CAS PubMed.
- C. Damm, R. Herrmann, G. Israel and F. W. Muller, Dyes Pigm., 2007, 74, 335–342 CrossRef CAS PubMed.
- C. Damm, D. Voltzke, H. P. Abicht and G. Israel, J. Photochem. Photobiol., A, 2005, 174, 171–179 CrossRef CAS PubMed.
- C. Dong and X. Y. Ni, J. Macromol. Sci., Part A: Pure Appl. Chem., 2004, 41, 547–563 CrossRef.
- M. Schmitt, S. Kuhn, M. Wotocek and R. Hempelmann, Z. Phys. Chem., 2011, 225, 297–311 CrossRef CAS.
- M. Schmitt, Macromol. Chem. Phys., 2012, 213, 1953–1962 CrossRef CAS PubMed.
- E. R. Carraway, A. J. Hoffman and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 786–793 CrossRef CAS PubMed.
- S. Moribe, T. Ikoma, K. Akiyama, Q. W. Zhang, F. Saito and S. Tero-Kubota, Chem. Phys. Lett., 2007, 436, 373–377 CrossRef CAS PubMed.
- M. Schmitt, Analyst, 2013, 138, 12 RSC.
- J. M. Coronado, A. J. Maira, J. C. Conesa, K. L. Yeung, V. Augugliaro and J. Sorie, Langmuir, 2001, 17, 5368–5374 CrossRef CAS.
- C. P. Kumar, N. O. Gopal, T. C. Wang, M. S. Wong and S. C. Ke, J. Phys. Chem. B, 2006, 110, 5223–5229 CrossRef CAS PubMed.
- P. A. van Hal, M. P. T. Christiaans, M. M. Wienk, J. M. Kroon and R. A. J. Janssen, J. Phys. Chem. B, 1999, 103, 4352–4359 CrossRef CAS.
- O. I. Micic, Y. N. Zhang, K. R. Cromack, A. D. Trifunac and M. C. Thurnauer, J. Phys. Chem., 1993, 97, 7277–7283 CrossRef CAS.
- T. Kawano and H. Imai, Cryst. Growth Des., 2006, 6, 1054–1056 CAS.
- M. Schmitt, R. Schulze-Pillot and R. Hempelmann, Phys. Chem. Chem. Phys., 2011, 13, 690–695 RSC.
- M. Schmitt, Macromol. Chem. Phys., 2011, 212, 1276–1283 CrossRef CAS PubMed.
- M. Schmitt and F. Heib, RSC Adv., 2014, 4, 17639–17647 RSC.
- ASTM, ASTM G173-03, ASTM International, 2012, DOI:10.1520/G0173-03E01.
- S. C. Liufu, H. N. Xiao and Y. P. Li, Mater. Lett., 2005, 59, 3494–3497 CrossRef CAS PubMed.
- G. Socrates, Infrared characteristic group frequencies “Table and Charts”, Wiley, 1994 Search PubMed.
- A. B. Djurisic and Y. H. Leung, Small, 2006, 2, 944–961 CrossRef CAS PubMed.
- D. W. Bahnemann, C. Kormann and M. R. Hoffmann, J. Phys. Chem., 1987, 91, 3789–3798 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- M. Schmitt, RSC Adv., 2014, 4, 1907–1917 RSC.
- A. Degen and M. Kosec, J. Am. Ceram. Soc., 2003, 86, 2001–2010 CrossRef CAS PubMed.
- D. Bahnemann, A. Henglein and L. Spanhel, Faraday Discuss., 1984, 151–163 RSC.
- S. C. Liufu, H. N. Mao and Y. P. Li, J. Colloid Interface Sci., 2005, 281, 155–163 CrossRef CAS PubMed.
- K. Kobayakawa, Y. Nakazawa, M. Ikeda, Y. Sato and A. Fujishima, Ber. Bunsen-Ges.-Phys. Chem., 1990, 94, 1439–1443 CrossRef CAS PubMed.
- Y. Oosawa and M. Gratzel, J. Chem. Soc., Chem. Commun., 1984, 1629–1630 RSC.
- Y. Oosawa and M. Gratzel, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 197–205 RSC.
- G. Ramakrishna and H. N. Ghosh, Langmuir, 2003, 19, 3006–3012 CrossRef CAS.
- Z. L. S. Seow, A. S. W. Wong, V. Thavasi, R. Jose, S. Ramakrishna and G. W. Ho, Nanotechnology, 2009, 20 Search PubMed.
- F. Mahdavi, T. C. Bruton and Y. Z. Li, J. Org. Chem., 1993, 58, 744–746 CrossRef CAS.
- A. A. Vinokurov, L. E. Derlyukova and V. V. Ganin, Russ. Chem. Bull., 1993, 42, 1137–1140 CrossRef.
- Z. R. R. Tian, J. A. Voigt, J. Liu, B. McKenzie, M. J. McDermott, M. A. Rodriguez, H. Konishi and H. F. Xu, Nat. Mater., 2003, 2, 821–826 CrossRef CAS PubMed.
- A. B. Djurisic, W. C. H. Choy, V. A. L. Roy, Y. H. Leung, C. Y. Kwong, K. W. Cheah, T. K. G. Rao, W. K. Chan, H. T. Lui and C. Surya, Adv. Funct. Mater., 2004, 14, 856–864 CrossRef CAS PubMed.
- C. G. Tian, B. D. Mao, E. B. Wang, Z. H. Kang, Y. L. Song, C. L. Wang and S. H. Li, J. Phys. Chem. C, 2007, 111, 3651–3657 CAS.
- W. Y. Choi, A. Termin and M. R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1994, 33, 1091–1092 CrossRef PubMed.
- H. Yamashita, Y. Ichihashi, M. Takeuchi, S. Kishiguchi and M. Anpo, J. Synchrotron Radiat., 1999, 6, 451–452 CrossRef CAS PubMed.
- M. Anpo, Bull. Chem. Soc. Jpn., 2004, 77, 1427–1442 CrossRef CAS.
- M. Anpo, Pure Appl. Chem., 2000, 72, 1265–1270 CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- F. Weichelt, R. Emmler, R. Flyunt, E. Beyer, M. R. Buchmeiser and M. Beyer, Macromol. Mater. Eng., 2010, 295, 130–136 CAS.
- H. M. Xiong, Z. D. Wang, D. P. Liu, J. S. Chen, Y. G. Wang and Y. Y. Xia, Adv. Funct. Mater., 2005, 15, 1751–1756 CrossRef CAS PubMed.
- V. Khrenov, M. Klapper, M. Koch and K. Mullen, Macromol. Chem. Phys., 2005, 206, 95–101 CrossRef CAS PubMed.
- S. X. Zhou, L. M. Wu, M. N. Xiong, Q. Y. He and G. D. Chen, J. Dispersion Sci. Technol., 2004, 25, 417–433 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Multiple additional figures and images concerning the synthesis, characterization, data evaluation, TEMs and ESR spectra are available free of charge. See DOI: 10.1039/c5nr00850f |
| ‡ Due to the superposition of the reflexes within the diffraction patterns (Fig. 3), the accuracy of the determined difference in the ratios is low. |
| This journal is © The Royal Society of Chemistry 2015 |