
Environmental photochemistry of single layered graphene oxide in water†
Yingcan
Zhao
and
Chad T.
Jafvert
*
Lyles School of Civil Engineering and Division of Environmental and Ecological Engineering, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: jafvert@purdue.edu; Tel: +1 (765)494 2196
First published on 2nd March 2015
Abstract
Graphene oxide (GO) is a carbonaceous nanomaterial that is a precursor material in the preparation of graphene, and because of its unique properties, it will likely be used in a number of industrial and consumer products in the future. Despite its name, it contains many epoxy, hydroxyl, and carboxyl functional groups on its edges and surface, making it easy to suspend in water. However, how it is transformed or mineralized in natural aquatic environments and its effects on natural processes within these environments remain largely unknown. Therefore, in this study, we report on the photochemical reactivity of single layered GO dispersed in water and irradiated with light within the solar spectrum that reaches the water bodies at the earth's surface (λ ≥ 300 nm). Upon irradiation, the visible color of a 5 mg L−1 GO suspension shifted from pale to dark brown, possibly indicating the repair of some of the π-bond structures; however, Raman spectroscopy indicated an increase in nonaromatic defects. To further examine how oxidation or reduction of the GO surface may occur upon solar light irradiation, we probed the production of various reactive oxygen species (ROS). By monitoring ROS production with selective and highly reactive chemical probes, formation of superoxide anions (O2˙−), but not single oxygen (1O2) or hydroxyl radicals (·OH), was detected, indicating electron transfer from GO to dissolved molecular oxygen (O2). However, further electron transfer through reduction of O2˙− did occur, as hydrogen peroxide (H2O2) was found to accumulate, forming 3 μM H2O2 in a suspension of 5 mg L−1 GO after 4 hours of irradiation.
Nano impactIn addition to fullerenes and carbon nanotubes, graphene and graphene oxide are major types of carbon-based nanomaterials. As a precursor material in the preparation of graphene, and because of its unique properties, graphene oxide (GO) will likely be used in a number of industrial and consumer products in the future. The types of products in which it will find application will partially depend on whether inclusion within these products contributes to human and environmental exposure, and the degrees of risk associated with these exposures. Because it contains many hydrophilic functional groups, it is easy to suspend in water. Hence, if it is released to the environment, exposure is likely to occur in aquatic environments. Yet, very limited research has been conducted regarding the environmental fate of GO, its transport mechanisms in the environment, or its toxicity to aquatic species. As a result, this study investigates the photochemical reaction of GO as it is one of the more likely fate processes acting on GO in aquatic environments. This study provides evidence that GO is chemically altered upon irradiation with light within the solar spectrum and that GO serves as an electron donor, transferring electrons to molecular oxygen to form reactive oxygen species (including superoxide anions and hydrogen peroxide). This type of information is critical for assessing potential impacts of graphene oxide on aquatic environments. |
Introduction
Many efforts are under way to discover new ways to use graphene materials and their derivatives in electronic devices, for drug delivery, in biosensors, in solar energy conversion, as catalysts, and in many other types of applications.1–7 Due to the variety of industrial sectors where manufactured graphene materials may find application and due to expected future production rates, release of and exposure to graphene-based materials in natural and built environments may be anticipated, raising concerns over the potential negative effects on human and ecosystem health if precautions are not taken to control exposure. Unfortunately, knowledge regarding the fate, transport, and toxicity of graphene and its derivatives is currently limited, especially with respect to aquatic environments. Graphene oxide (GO) is a precursor material in the preparation of graphene and, despite its name, it has on its surface several different types of functional groups, including epoxy, hydroxyl, and carboxyl groups.8 The presence of these functional groups on GO makes it hydrophilic and easy to disperse in water.9Because it is easy to disperse in water, GO has been studied both as a potential aquatic pollutant and as a potential material to selectively kill cancer cells due to its toxicity to human cells.6,10 For example, Liao et al. observed that sonicated (smaller) GO exhibited greater hemolytic activity to human cells compared to larger GO materials. Viability assay experiments revealed that both graphene and GO have toxic effects on human skin fibroblasts.11 Another research group reported that single-layer GO had dose-dependent toxicity to human lung epithelial cells and fibroblasts and caused obvious toxicity when doses were above 50 mg L−1, indicating risk to human health.12 Hu et al. found that GO could disrupt cell membranes by direct interactions occurring between cell membranes and GO nanosheets.13 In contrast, another research group used different sized GO and found that all the different sized materials showed no toxicity to A549 cells, which are typical human lung cells.14 The variability among the results might be attributed to the various methods by which GO and graphene were produced or suspended in water. However, there is little doubt that GO might cause some toxicological effects on humans, motivating further studies on the fate and effects of GO in the natural environment. Environmental effects may include toxicity to micro-organisms and other organisms further up the food chain. Indeed, Akhavan et al. showed that hydrazine-reduced GO was more toxic to bacteria than the parent GO, and suggested that this resulted from the “sharper” nanowalls on the reduced GO (RGO).15
Because the disrupted π-bond structure in GO absorbs a significant amount of light within the solar spectrum, environmental fate processes acting on GO are expected to include photochemical processes. Indeed, it is well known that photo-irradiation is a good method for “reducing” GO, at least with lamp light that occurs in UV regions which may or may not occur solely within the solar spectrum.16 For example, in a study reporting on photo-reduction of GO conducted by Matsumoto et al.,16 experiments were performed using a xenon lamp of unknown spectral output, although this type of lamp generally emits light down to 280 nm, approximately 20 nm below the spectral limit of solar light measured at the earth's surface. Matsumoto et al., however, did measure CO2 and H2 generation from aqueous GO suspensions and noted a drastic decrease in H2 production when xenon lamp light was filtered through a 390 nm cutoff filter. While not reported, it seems likely that a similar decrease in CO2 production would occur under a 390 nm cutoff filter. It is somewhat interesting that while the overall reaction can be termed photo-reduction of GO, it is not clear if any of the remaining carbon in GO has actually been reduced, as the simple loss of CO2 from the carboxyl groups of GO is the result of a rearrangement reaction, where the carbon–carboxylic acid bond is broken and replaced with a carbon–hydrogen bond. Hence, although the average oxidation state of carbon in GO is lower, it may result from the loss of carbon that was already highly oxidized, as previously reported to occur during photolysis of carboxylated multi-walled carbon nanotubes,17 and not from any oxidation reaction of specific carbon atoms in GO. Similar to the lack of information in the literature regarding photochemical carbonaceous products, there is a general lack of information on the generation of reactive oxygen species (ROS) by GO under solar light. Hou et al.18 irradiated an aqueous dispersion of single layer GO with light not strictly within the solar spectrum (at energies in the range 3.94–4.43 eV; i.e., at λ = 280–315 nm) and, similar to Matsumoto et al.,16 noted that GO became visibly darker over the irradiation period, but suggested through XPS analysis that this occurred due to the loss of hydroxyl groups through hemolytic removal of ·OH groups and formation of more conjugated π-bonds, rather than through decarboxylation alone. Further, through electron paramagnetic resonance spectroscopy, dose-dependent exponential growth in radical production on the GO surface was shown to occur, presumably after the loss of ·OH; however, it was reported that ·OH was not observed, and the methodology of ·OH detection was not reported.
For other carbon-based nanomaterials, several reactive oxygen species have been shown to be generated under sunlight conditions (λ > 300 nm). For example, singlet oxygen (1O2) was generated in aqueous suspensions of C60 clusters (i.e., nano-precipitates), oxidizing C60 to more polar water-soluble products.19–21 Aqueous dispersions of carboxylated and PEG-functionalized single walled carbon nanotubes (SWCNTs) produced a significant amount of ROS, including singlet oxygen (1O2), superoxide anions (O2˙−), and hydroxyl radicals (·OH).22,23 As a result, in this study, the ability of aqueous dispersions of single-layered GO to generate reactive oxygen species (ROS) upon exposure to light within the solar spectrum (λ = 300–410 nm) was investigated. Based on the experimental results of this study and the previous work cited above, a mechanism for ROS generation by photosensitized GO in water is proposed. Information on ROS generation during solar irradiation is significant not only to evaluate the ecological risks associated with GO, but also to better understand the transformation pathways of carbon within GO.
Materials and methods
Materials
An aqueous dispersion of single layer graphene oxide (GO), synthesized by a modified Hummers method, was purchased from Advanced Chemicals Supplier (ACS) Material, LLC (Medford, MA), and used as-received. According to the manufacturer, the material is approximately 80% single-layer GO (with the remainder being multi-layered) and is in the size range of 0.5 to 2.0 μm, with a thickness of 0.6 to 1.2 nm. A previous study reported the presence of phenolic hydroxyl, carboxylic, and epoxy groups within the structure of this particular graphene oxide.24 More information on the GO material is provided in the ESI† (Fig. S1). Furfuryl alcohol (FFA), 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT), p-chlorobenzoic acid (pCBA), N,N-diethyl-p-phenylenediamine hemioxalate (DPD), horseradish peroxidase (HRP), and superoxide dismutase (SOD) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of the highest purity and were used as-received. All aqueous samples were prepared with water purified using a Barnstead Nanopure system (Dubuque, IA) after R/O pretreatment.Preparation of aqueous graphene oxide dispersions
A stock GO dispersion (10 mg mL−1) was diluted with water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100), and sonicated under low energy (8890R-MT ultrasonic bath from Cole-Parmer, Vernon Hills, IL) for 2 hours to produce a uniform dispersion of 100 mg L−1 GO. When not in use, the GO suspensions were stored in the dark at room temperature. In most experiments, samples were buffered to pH 7 with a final phosphate buffer concentration of 5 mM. Buffers were prepared with phosphate salts (i.e., KH2PO4 and K2HPO4).
:100), and sonicated under low energy (8890R-MT ultrasonic bath from Cole-Parmer, Vernon Hills, IL) for 2 hours to produce a uniform dispersion of 100 mg L−1 GO. When not in use, the GO suspensions were stored in the dark at room temperature. In most experiments, samples were buffered to pH 7 with a final phosphate buffer concentration of 5 mM. Buffers were prepared with phosphate salts (i.e., KH2PO4 and K2HPO4).
Irradiation and GO analysis
For all irradiation experiments, samples were placed in a series of borosilicate glass tubes sealed with PTFE-lined caps and exposed to sixteen 24 W black-light phosphor lamps (RPR-3500 Å lamps from Southern New England Ultraviolet, Branford, CT) that emit light from 300 to 400 nm, with the maximum intensity occurring near 350 nm. A figure comparing the spectral output of these lamps to that typically found for solar light at the earth's surface is provided in the ESI† (Fig. S2). All samples were irradiated in a Rayonet merry-go-round photochemical reactor that rotated the samples at 5 rpm to ensure uniform light exposure of all samples over the irradiation period. Dark control samples were prepared at the same time as irradiated samples, and were wrapped with aluminum foil and kept in a dark environment over the same time period. All experiments were performed in duplicate or triplicate. At specific times during the irradiation period, the irradiated samples and the dark control samples were recovered for analysis. The UV-visible light absorbance spectra were recorded with a Varian Cary 50 UV/Vis spectrophotometer using quartz cuvettes with 1 cm path length. The Raman spectra of the samples were monitored by using an excitation wavelength of 633 nm, and scanning from 1000–3000 cm−1 to obtain information on the characteristic D, G and 2D band intensities of GO. For all Raman spectra measurements, irradiated and dark control samples were prepared and analyzed in the absence of phosphate buffers.ROS measurement
Reactive oxygen species (ROS) were detected by using specific chemical probes that selectively react with each ROS at near diffusion-limited rates. Only one probe was used at a time, as those for singlet oxygen (1O2) superoxide anions (O2˙−), and hydroxyl radicals (·OH) require that they be added before the irradiation period, as the measured response is due to accumulative ROS production; whereas analysis of hydrogen peroxide (H2O2) was performed on samples by adding the necessary reagents after irradiation. 1O2 was monitored by the loss of FFA.25 Detailed information about detection of 1O2 with FFA is included in the ESI.† Previously, we used a nitro blue tetrazolium salt (referred to as NBT2+) as a scavenger for O2˙− that was produced upon solar light irradiation of functionalized carbon nanotubes (CNTs).22 However, in the presence of NBT2+, GO was found to flocculate and settle; hence experiments using XTT as a scavenging probe for O2˙− were attempted with success. The reaction between XTT and O2˙− leads to the formation of a soluble product that was detected spectrophotometrically at 470 nm after filtering the samples through 0.2 μm membrane filters to remove GO, which also absorbs light at 470 nm. To confirm that transformation of XTT was through reaction with O2˙−, in some GO + XTT samples, superoxide dismutase (SOD) was added, as it significantly decreases the steady-state concentration of O2˙− by enzymatically catalyzing its disproportionation to hydrogen peroxide (H2O2), confirming that transformation of XTT was due to reaction with O2˙−. p-Chlorobenzoic acid (pCBA) was added to some samples to determine if ·OH was generated during irradiation.22pCBA reacts rapidly with ·OH resulting in the loss of pCBA from the system; the complete pCBA method is described in the ESI.† H2O2 temporal concentrations were measured by horseradish peroxidase (HRP) catalyzed oxidation of DPD, with HRP and DPD added to the samples after irradiation or incubation in the dark.26,27 Before analyzing the samples for H2O2, irradiated and dark control samples were filtered through 0.2 μm membrane filters to remove GO, which otherwise would interfere with DPD spectrophotometric detection. After filtration, 1 mL of each sample was added to 1 mL of phosphate buffer (pH 6), followed sequentially by addition of 50 μL of 30 mM DPD solution and 50 μL of 30 U mL−1 HRP. H2O2 concentrations were determined by comparing the sample absorbance values at 551 nm to those of known standards (i.e., from a standard curve).Results and discussion
Upon irradiation of an aqueous dispersion of single layered GO with light within the solar spectrum, the GO dispersion becomes visibly darker as shown in Fig. 1a. This is consistent with previous observations noted in the introduction section that reported on exposure of GO to shorter wavelength UV light.16,18 Similarly, light attenuation (Fig. 1b) of the GO suspension increased across the whole spectrum from 220 to 820 nm. Note that although the figure reports “absorbance”, the response is due to both light absorbance and scattering because the samples are nanoparticle suspensions that not only absorb light within this wavelength range but also scatter light, with presumably much of the light attenuation at longer wavelengths occurring due to light scattering. The GO spectra before irradiation do exhibit two characteristic peaks: a maximum peak at approximately 230 nm, which corresponds to π → π* transition of aromatic C–C bonds, and a shoulder peak at 303 nm, which is due to n → π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.28 Over the course of a 2 h irradiation period, the absorption peak at 230 nm was gradually red-shifted to approximately 255 nm and the shoulder peak at 303 nm disappeared, potentially indicating that some of the electronic conjugations within the graphene sheets were restored as previously suggested upon reduction of GO with light at shorter wavelengths.18,29
O bonds.28 Over the course of a 2 h irradiation period, the absorption peak at 230 nm was gradually red-shifted to approximately 255 nm and the shoulder peak at 303 nm disappeared, potentially indicating that some of the electronic conjugations within the graphene sheets were restored as previously suggested upon reduction of GO with light at shorter wavelengths.18,29
 | ||
| Fig. 1 (a) Photograph of 5 mg L−1 GO at pH 7, before (left vial) and after (right vial) irradiation for 2 hours with lamps that emit light from 300 to 410 nm, and (b) the change in the UV-visible light absorption spectra of similar GO suspensions with increasing time of irradiation. | ||
Raman spectroscopy is often used to probe the differences in the electronic properties of carbon nanomaterials.30Fig. 2 shows the Raman spectral changes in GO as a function of irradiation time. The spectra show D, G, and 2D bands, which are three characteristic peaks of GO. The G band occurs at 1580 cm−1 which results from the vibration of sp2-bonded carbon, and is an indication of the relative extent of aromaticity. The D band at 1350 cm−1 is assigned to the vibration of sp3 carbon atoms (i.e., nonaromatic carbon). The relative intensity, or the ratio of the D-to-G band intensities (ID/IG), is often used as a qualitative measure for the degree of disorder caused by nonaromatic sp3 carbon defects that often occur at the edges or as ripples or holes within the GO structure.31Fig. 2 shows that after 24 h of irradiation, the ID/IG ratio increased from 0.45 (0 h) to 0.68 (24 h). This increase suggests an increase in the number of defects (e.g., functionalized carbon) on the already functionalized graphene oxide sheets. These defects may be sites for ROS production, which is discussed subsequently.
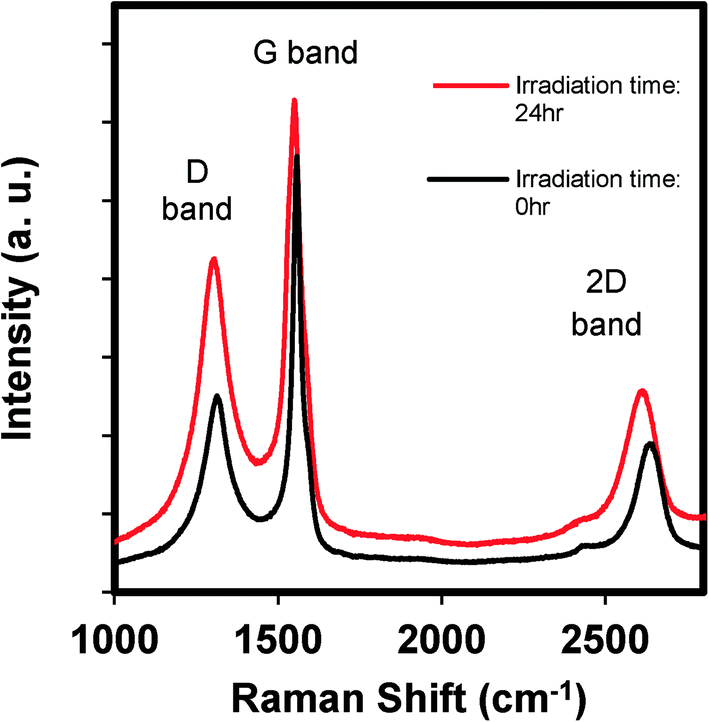 | ||
| Fig. 2 The Raman spectra of GO before and after irradiating an aqueous GO suspension (100 mg L−1) for 24 hours, where the spectra have been normalized to the intensity of the G band. | ||
By monitoring ROS production with selective and highly reactive chemical probes, formation of O2˙−, but not 1O2 or ·OH, was detected. Irradiated samples containing furfuryl alcohol as a scavenging probe for 1O2 showed no decay in furfuryl alcohol after 24 h of irradiation (Fig. S3†). However, in GO suspensions containing XTT, a significant increase in light absorbance at 470 nm occurred upon irradiation and after filtering out GO after irradiation over time. This increase in absorbance occurs when the reaction product between XTT and O2˙− has an absorbance maximum22 (Fig. 3). Fig. 3a also indicates that addition of SOD almost completely inhibited XTT reduction, further suggesting that XTT product formation was caused directly by the reaction of XTT with O2˙− as SOD rapidly converts O2˙− to H2O2 through a disproportionation reaction, reducing the amount of the XTT product formed. In the absence of XTT, the irradiated and dark control GO samples showed little change in absorbance at 470 nm over the same time period. Note that because the light absorption spectral changes shown in Fig. 3b (and reported at 470 nm in Fig. 3a) are on samples filtered through 0.2 μm membrane filters (i.e., after removal of GO), the increase in absorbance at 470 nm is due only to XTT product formation and not to the increase in light absorbance caused by GO on unfiltered samples, as shown in Fig. 1. An image showing the course of the reaction from 0 to 3 hours for a 5 mg L−1 GO suspension containing 0.1 mM XTT, prior to filtration, is provided in the ESI† (Fig. S4). Even with the increase in the overall absorbance caused by GO, the pink product of the reaction between O2˙− and XTT is evident.
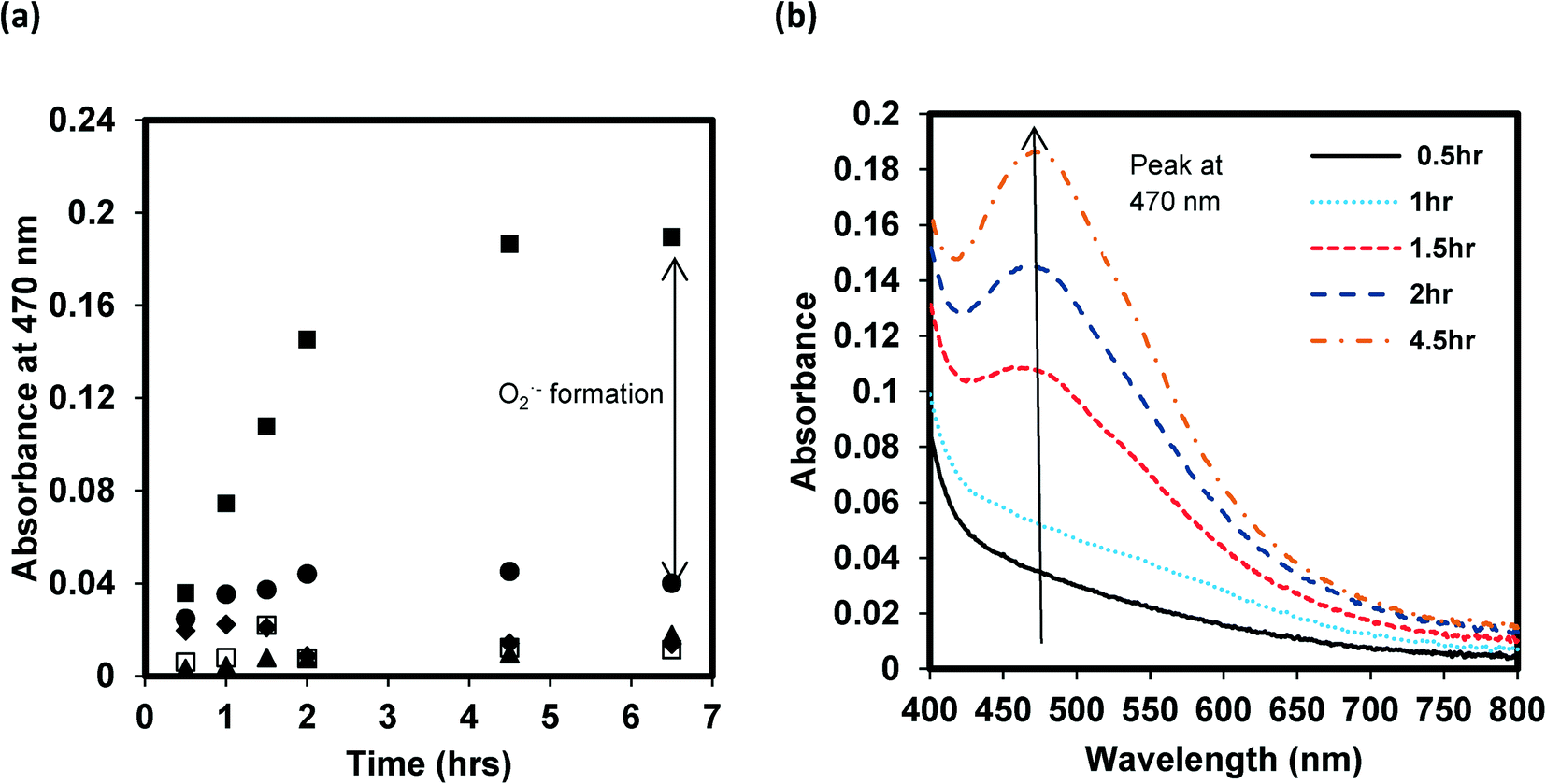 | ||
| Fig. 3 (a) Evidence of O2˙− formation by reaction with XTT, forming the pink colored reduction product that absorbs light at λmax = 470 nm, upon lamp light irradiation of a 5 mg L−1 GO suspension at pH 7. The symbols represent samples containing XTT (0.1 mM) alone (▲); GO (5 mg L−1) alone (✦); GO (5 mg L−1) and XTT (0.1 mM) (■); GO (5 mg L−1), XTT (0.1 mM) and SOD (40 U mL−1) (●); and the corresponding dark control sample of GO (5 mg L−1) and XTT (0.1 mM) (□). (b) The change in the UV-visible light absorption spectra of a suspension of GO (5 mg L−1) and XTT (0.1 mM) as a function of irradiation time, after filtering the samples through 0.2 μm filters to remove GO. | ||
With significant O2˙− formation, there is a high probability that hydrogen peroxide will form also, and potentially accumulate in solution. As noted above, H2O2 can be formed by disproportionation of O2˙− with enzymes such as SOD, accelerating the rate of the reaction considerably. As the disproportionation reaction suggests, conversion can occur also through transfer of another electron to the protonated form of the superoxide anion, HO2· (hydroperoxyl radical), which upon electron transfer extracts a proton from solution to form H2O2. Whether it occurs by disproportionation or another electron transfer process, both electrons must originate from GO in the absence of an additional electron donor. Hence, accumulation of H2O2 was measured by using the DPD–horseradish peroxidase (HRP) assay, with DPD/HRP added to the aqueous samples after irradiating the GO dispersions, and then after removing GO by filtration. Although H2O2 is somewhat reactive in this system, it has a much longer half-life than the other ROS, even under solar light irradiation. Fig. 4a clearly shows that the H2O2 concentration in the GO dispersions increased over an irradiation period of 4 h, whereas no increase occurred in the dark control samples or in the absence of GO. The H2O2 concentrations shown in Fig. 4a are calculated from the sample absorbance values measured at 551 nm after filtration, using the H2O2 standard curve shown in Fig. 4b. Hence, the initial values at time zero of 0.2–0.5 μM are due to absorbance reading at or below 0.005, and likely due to trace contamination resulting in a small positive interference. Despite this, the results indicate that H2O2 was indeed produced and accumulated during irradiation of 5 mg L−1 GO with light within the solar spectrum, accumulating to over 3 μM after an irradiation period of 4 h. Assuming a carbon content of 80% in the original GO, the 5 mg L−1 GO concentration translates to a molar concentration of 3 mM carbon. Hence, over the 4 h irradiation period, approximately 1 molecule of H2O2 was produced for every 1000 carbon atoms in GO.
 | ||
| Fig. 4 (a) Evidence of the increase in H2O2 concentration upon lamp light irradiation of GO at pH 7 over time by reaction of the accumulated H2O2 with DPD through a HRP catalyzed reaction, producing the red colored DPD Würster dye product that absorbs light at 551 nm. All samples contain DPD and HRP, added after irradiating samples which contained 5 mg L−1 GO (■) or pure water (▲), or after incubating samples in the dark which contained 5 mg L−1 GO (□); DPD and HRP were added after filtering out GO through 0.2 μm membrane filters. (b) The standard curve of H2O2 obtained using a DPD/HRP method. | ||
Although it is known that sunlight can cleave the oxygen–oxygen bond in H2O2 to form ·OH, the reaction is slow.23,32 Alternatively, ·OH may be formed more rapidly if transfer of an additional electron from GO to H2O2 occurs, as was found to be the case for carboxylated single walled carbon nanotubes under solar irradiation.23 In order to determine whether there are hydroxyl radicals produced by GO, pCBA was added to some samples, as it rapidly scavenges ·OH resulting in the loss of pCBA. However, no pCBA decay was observed for both irradiated and dark control samples over the 24 hour time period of the experiments (see the ESI,† Fig. S5), suggesting that a negligible amount of ·OH was produced or that ·OH scavenging by GO was rapid and significant, reducing its pseudo-steady-state concentration. Although scavenging of ·OH by GO is likely to occur, as the initial electron transfer that results in its formation would occur at the GO surface such that the site of its generation would be in close proximity to GO π-bonds at which it could be consumed, it is also likely that not much is produced, otherwise the concentration of its precursor, H2O2, would not accumulate to such a degree, and the chromophore content of GO would not be enhanced as irradiation proceeds.
Conclusions
In summary, when exposed to light within the solar spectrum, aqueous dispersions of single layered GO do become darker, indicating an increase in chromophore content or at least an increase in absorptivity of the existing chromophores in GO, yet Raman spectroscopy indicates an increase in nonaromatic defects. Clearly, upon exposure to light, electron transfer reactions occur from GO to O2, forming O2˙− and significant quantities of H2O2 as depicted in Scheme 1, and because these are both reduction reactions, this must result in the overall oxidation of GO carbon. Although it is likely that some of the generated ROS react directly with the GO surface, this clearly does not occur stoichiometrically, as evidenced by the buildup of H2O2 concentration over a time period of several hours. These results suggest that future studies should examine whether these electron transfer reactions are responsible for some of the toxicological effects observed for GO. In a recent study,27 we have reported that electrons from common biological reducing agents (i.e., NADH) can be shuttled through single-walled carbon nanotubes to molecular oxygen in the dark, resulting in ROS production and DNA cleavage. It is likely that a similar mechanism resulting in oxidative stress may occur also in the case of GO, but this hypothesis is yet to be tested. | ||
| Scheme 1 Proposed pathway for ROS production by photosensitization of GO in water. | ||
Acknowledgements
This research was supported by the United States Environmental Protection Agency (U.S. EPA) STAR grant program (grant no. RD 83485801). We also acknowledge and thank the support from the China Scholarship Council (CSC) through a Ph.D. fellowship awarded to Yingcan Zhao to study at Purdue University. This work was presented at the 9th International Conference on the Environmental Effects of Nanoparticles and Nanomaterials in South Carolina, USA, in September 2014.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- J. H. Jung, D. S. Cheon, F. Liu, K. B. Lee and T. S. Seo, Angew. Chem., Int. Ed., 2010, 49, 5708–5711 CrossRef CAS PubMed.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS PubMed.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- F. Barroso-Bujans, S. Cerveny, R. Verdejo, J. del Val, J. Alberdi, A. Alegría and J. Colmenero, Carbon, 2010, 48, 1079–1087 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, B. Tian, Y. Zhang and Z. Liu, Biomaterials, 2012, 33, 2206–2214 CrossRef CAS PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6 Search PubMed.
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef CAS PubMed.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- Y. Matsumoto, M. Koinuma, S. Ida, S. Hayami, T. Taniguchi, K. Hatakeyama, H. Tateishi, Y. Watanabe and S. Amano, J. Phys. Chem. C, 2011, 115, 19280–19286 CAS.
- J. L. Bitter, J. Yang, S. BeigzadehMilani, C. T. Jafvert and D. H. Fairbrother, Environ. Sci.: Nano, 2014, 1 Search PubMed.
- X.-L. Hou, J.-L. Li, S. C. Drew, B. Tang, L. Sun and X.-G. Wang, J. Phys. Chem. C, 2013, 117, 6788–6793 CAS.
- W.-C. Hou and C. T. Jafvert, Environ. Sci. Technol., 2008, 43, 362–367 CrossRef.
- W.-C. Hou and C. T. Jafvert, Environ. Sci. Technol., 2009, 43, 5257–5262 CrossRef CAS.
- W.-C. Hou, L. Kong, K. A. Wepasnick, R. G. Zepp, D. H. Fairbrother and C. T. Jafvert, Environ. Sci. Technol., 2010, 44, 8121–8127 CrossRef CAS PubMed.
- C.-Y. Chen and C. T. Jafvert, Environ. Sci. Technol., 2010, 44, 6674–6679 CrossRef CAS PubMed.
- C.-Y. Chen and C. T. Jafvert, Carbon, 2011, 49, 5099–5106 CrossRef CAS PubMed.
- H. Chen, B. Gao and H. Li, J. Hazard. Mater., 2015, 282, 201–207 CrossRef CAS PubMed.
- W. R. Haag and J. Hoigne, Environ. Sci. Technol., 1986, 20, 341–348 CrossRef CAS PubMed.
- H. Bader, V. Sturzenegger and J. Hoigne, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
- H.-S. Hsieh, R. Wu and C. T. Jafvert, Environ. Sci. Technol., 2014, 48, 11330–11336 CrossRef CAS PubMed.
- J. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- J. G. Radich and P. V. Kamat, ACS Nano, 2013, 7, 5546–5557 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- W.-C. Hou, S. BeigzadehMilani, C. T. Jafvert and R. G. Zepp, Environ. Sci. Technol., 2014, 48, 3875–3882 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4en00209a |
| This journal is © The Royal Society of Chemistry 2015 |