
Behaviour and mobility of U and Ra in sediments near an abandoned uranium mine, Cornwall, UK†
Saifeldin M.
Siddeeg
*abc,
Nicholas D.
Bryan
d and
Francis R.
Livens
a
aCentre for Radiochemistry Research, School of Chemistry, The University of Manchester, Manchester, M13 9PL, UK. E-mail: saifeldin.siddeeg@manchester.ac.uk
bCollege of Science, Department of Chemistry, King Khalid University, P.O. Box 9004, Abha, Saudi Arabia
cChemistry and Nuclear Physics Institute, Atomic Energy Commission, P.O. Box 3001, Khartoum, Sudan
dNational Nuclear Laboratory, 5th Floor, Chadwick House, Warrington Road, Birchwood Park, Warrington, WA3 6AE, UK
First published on 3rd December 2014
Abstract
Sediment samples were collected from the vicinity of the abandoned South Terras uranium mine in south-west UK and analysed for uranium and 226Ra to explore their geochemical dispersion. The radioactivity concentrations in the sediment samples were measured using alpha spectrometry for uranium, and gamma spectrometry for radium. Sequential chemical extraction was applied to selected sediments in order to investigate the speciation of the radionuclides and their association with stable elements. The activity ratio of the uranium isotopes was used to explore the mobility of uranium, and scanning electron microscopy (SEM) and electron microprobe analysis (EMPA) were used to characterise the sediments. The radiochemical results identified two locations with enhanced radioactivity, so two samples from these locations were further investigated. The geochemical distribution of the radionuclides in these two samples varies within the five operationally-defined fractions. In one sample, the majority of the uranium was released from the ‘carbonate’ fraction, followed by the organic fractions. Similarly, in the second sample, the uranium was mainly resealed from the carbonate fraction, although a considerable percentage associated with the resistant fraction. The fractionation trend of radium noticed to show some similarities to that of barium, as expected from the similarity in their chemistries. Geochemical distributions of the stable elements, such as Mn, Ti and As, were different in the enhanced radioactivity samples. The activity ratio of 234U/238U shows different trends in the two sediments, signifying the impact of organic matter and/or the exchange between water and sediment. SEM and EMPA analysis identified uranium-bearing phases in association with potassium, calcium, iron, manganese and arsenic.
Environmental impactDifferent compartments, such as water, food and air, if contaminated can affect human health. Following the industrial revolution, the relationship between local geochemistry and actual intake of chemicals play a vital role in mobility of contaminants in the environment. As uranium present a chemical and radiological hazard, it is important to investigate its distribution in specific locations such as former uranium mining areas. The study targets South Terras abandoned uranium mine, Cornwall, UK as a natural analogue to explore the fate of natural radionuclides. Uranium and radium have been geochemically characterised in sediments collected from the vicinity by different techniques. The work will be of interest to a wide range of the scientific community, including environmental chemists, radiochemists and geochemists. |
1. Introduction
Natural decay series radionuclides show elevated concentrations in all igneous rocks.1 Therefore, regions with acid igneous rocks, such as granite, are often of interest as potential uranium mining areas. Following mining and milling, large amounts of waste containing isotopes from the uranium decay chain are produced and dumped in spoil heaps and mill tailings.2 The tailings left behind hold most of the radium from the ore, as a co-precipitate with barium or lead, usually associated with fine grained material in the waste. Alpha emitters, such as 238U, 234U, 230Th and 226Ra, are the most important isotopes in the radiological assessment of many former uranium mining locations worldwide.3–6 Accordingly, continuous radiological surveillance of these sites is required, even after cessation, to monitor radioactivity in waste piles and spoil heaps.The south-west of the UK, with its scattered granitic intrusions, has a rich history of mining activities.7 In the context of radioactive deposits, the most significant mine in Cornwall is that at South Terras (50° 20.048′ N 4° 54.311′ W), which was the only UK mine worked primarily for uranium and, subsequently, radium. The potential for uranium contamination of water and sediment collected from the River Fal, which flows close to the vicinity of the mine, has been studied,8 and the results suggested that the uranium mine and spoil heaps at the mine were not significant sources of uranium in the river water. This was supported by a correlation found between the total cation concentrations and uranium in the surface water, thereby suggesting that uranium in the river water originated from rock weathering. Nevertheless, the concentration of uranium locally in sediment beneath the outflow pipe was relatively high, and reaching up to 1000 ppm.
Nearly a century after the cessation of mining activities in South Terras, the spoil heaps can be used as a natural laboratory in which to study the environmental geochemistry of the natural radionuclides. The area is of particular interest due to the retention of the long-lived radionuclides and the presence of a variety of minerals and the associated stable metals.7 Therefore, surface sediments in the River Fal are of interest from a radiological and toxicological perspective.
The aim of this study is to use the disused uranium mine at South Terras to explore the behaviour of the natural radionuclides associated with the uranium mining activities, mainly 238U, 234U and 226Ra, in particular by means of:
• Quantifying the distribution of natural radionuclides associated with uranium mining, namely 238U, 234U and 226Ra.
• Characterising sediments with elevated radionuclide contents to investigate the geochemical associations and mobility of the radionuclides.
• Identifying factors affecting this transport, such as adsorption to organic matter, association with different geochemical phases within the sediments, relationship with trace elements.
2. Materials and methods
2.1 The study area and sampling
In the south-west region of the UK, Cornwall's high-temperature (300–500 °C) veins, oriented NE-SW and associated with diverse and complex mineralisation, have been exploited for different elements, including copper, tin, iron and lead. Other low-temperature veins (100–300 °C) cross the high-temperature veins. These contain a small amount of pitchblende, and have been explored for cobalt, nickel, iron, lead, uranium and then for radium.9 At South Terras, uranium was excavated mainly from pitchblende (U3O8) and uraninite (UO2) as primary ores, but secondary minerals, such as autunite [Ca(UO2)2(PO4)2·10H2O], zippeite [(UO2)3(SO4)2(OH)2·8H2O] and metatorbernite [Cu(UO2)2(PO4)2·8H2O], were reported to be common in the area.9The area around the abandoned mine is now covered by vegetation. It can be noticed that plants had grown in the mine debris and heaps of waste materials have been reshaped by erosion. Twenty surface sediments were collected by hands at a depth approximately at 5–10 cm beneath the surface water along an approximately 2 km stretch of the valley of the River Fal, running south from the South Terras mine site (Fig. 1). They were saved in Kraft envelopes for further analysis in the laboratory, where they were wet sieved through a 2 mm sieve and then air dried. Once dry, they were disaggregated using a mortar and pestle and homogenised to be suitable for chemical treatment.
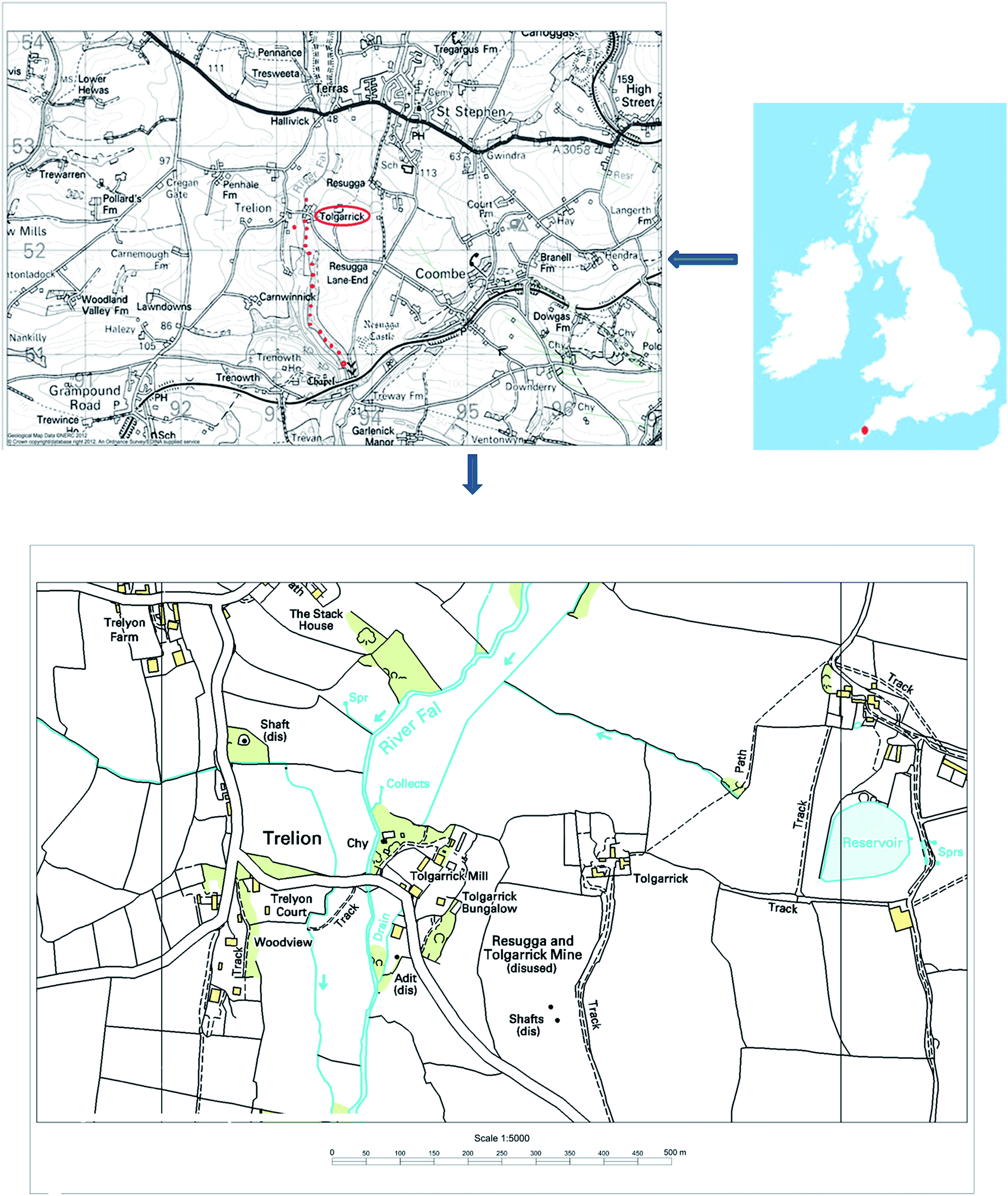 | ||
| Fig. 1 Cornwall map showing the sampling points along the river Fal. | ||
2.2 Physicochemical properties of the sediments
Organic matter (OM) content was estimated from loss on ignition.10 A porcelain crucible was ignited at 550 °C for 30 minutes in a muffle furnace, then allowed to cool in a desiccator and accurately weighed. From the bulk dry sediments, 1.0–2.0 g was placed in the crucible and weighed accurately, then transferred to a muffle furnace and heated to 550 °C for 5 hours. The hot crucible, containing the residue, was placed in the desiccator and cooled to ambient temperature. The crucible containing the ashed sediment was weighed accurately and the loss on ignition as a percentage was calculated.2.3 Mineral identification using X-ray diffraction
The dry sediments were sieved through 80 mesh to obtain the sediment fraction with 0.177 mm11 and a suitable amount (∼0.5 g) of each sample was placed on the sample holder. A smooth, flat surface was obtained using a glass slide, before placing the sample in the specimen position of the XRD. Mineral identifications were made using a Bruker D8 Advance diffractometer. The X-rays were generated from a Cu X-ray tube (Kα with a wavelength of 0.152 nm, current 30 mA at 40 kV) and the instrument is equipped with a standard scintillation detector. The scanning angle for the specimen was set from 5° to 75° with a step size of 0.02° s−1 and an exposure time of 30 minutes. Phase identification was performed using the Bruker Eva 14, version 2008 pattern analysis tool.2.4 Radioactivity content in sediments
An extraction chromatography column (UTEVA, 2.0 ml pre-packed column; Eichrom resin, Triskem, France) was preconditioned with 5 ml 3.0 M HNO3 before loading the solution. The beaker was washed with 5.0 ml 3.0 M HNO3 and the wash was passed through the column. Then the column was rinsed with consecutive additions of 5.0 ml of 3.0 M HNO3, 5.0 ml of 9.0 M HCl and 20.0 ml of 5.0 M HCl in 0.05 M H2C2O4. All these eluates were discarded and, finally, uranium was stripped with 15.0 ml of 1.0 M HCl. The eluent was evaporated to near dryness for electrodeposition in the presence of 1.0 ml 10% (w/v) KHSO4 using a heat lamp.
For uranium electroplating, 2.5 ml of 5 wt% NaHSO4, 2.0 ml of deionised water and 5.0 ml of 15.0 wt% Na2SO4 were added to the residue of the purified uranium fractions and heated gently until the residue dissolved. The solution was transferred to an electrodeposition cell and rinsed in with 3.0 ml deionised water, then 1.0 ml of 20.0 g L−1 (NH4)2C2O4 plating solution was added. The current was adjusted to 0.5 A for 5 minutes and then to 0.75 A for 90 minutes. One minute before the end, 2.0 ml of 25.0 wt% KOH was added and the power was turned off. The solution was discarded and the cell was washed with 2.0 ml 5.0 wt% NH4OH.
Finally, the stainless-steel disc was rinsed consecutively with a small volume of 5.0 wt% NH4OH, ethanol and acetone before being dried on a hotplate at 200 °C for 5 minutes.
The whole radiochemical separation method was validated using blank samples (deionised water spiked with the tracer) and the IAEA RM-314 reference material. The method was also tested using standard additions, in which a known amount of 238U was added to three duplicate samples and then the separation was performed.
2.5 Quality control
The analysis conducted, either for the total or the leached fraction (aqua regia), of the radionuclides was tested by regular quality control methods. The radiochemical separation was validated using blank solutions spiked with the tracer, standard additions and a standard reference material (IAEA-314). The blank analyses always gave less than 5 counts in each uranium region of interest, whereas all the sample analyses are based on signals of at least several hundred counts. In the standard additions, where a known amount of 238U was added to three duplicate samples and then the separation was performed on the two samples, the measured uranium recoveries were 92 ± 12%, 116 ± 17% and 87 ± 11% of the added uranium. The higher recovery with its counting error may indicate contamination and/or instrumentation error for this sample. Reference materials are used to verify the accuracy and overall performance of the analysis. They are chosen to closely match the matrix and analyte concentration of interest, so IAEA-314 sediment reference material was analysed for this purposes. The obtained results for the IAEA-314 were close to the recommended values, as can be seen in Table 1.| 226Ra Bq kg−1 | 238U mg kg−1 | |
|---|---|---|
| Measured | 774 ± 24 | 57.8 ± 0.9 |
| Recommended | 732 | 56.8 |
| 95% confidence interval | 678–787 | 52.9–60.7 |
2.6 Sequential chemical extraction
The method used in this study to determine the speciation of radionuclides in the sediments with the highest uranium content, S3 and S7, was the one that had been optimized for quantification of actinides in an organic-rich soil (approximately > 10%). The only modification was using aqua regia instead of the strong acids (HClO4/HF) to avoid complete oxidation of organic matter and dissolution of silica, because the interest is to examine radionuclide and heavy metal mobility rather than obtaining the total concentrations in the residual fraction.All reaction steps were performed in duplicate in 50.0 ml polyethylene centrifuge tubes with a solid/reagent ratio of 1.0 g/15.0 ml.14 The reagents and conditions for each step are given in ESI (Table S1†). At the beginning of the method, 1.0 g sample was wetted overnight with water before conducting the extraction steps. To separate the extract solution following each extraction step, the samples were filtered through 0.22 μm filter and centrifuged at 4500 rpm/20 minutes. The solid residue was saved for later steps and uranium and radium were determined in the solutions. To the solutions, tracers 232U (∼70 y) for uranium and 225Ra (∼15 d), in equilibrium with the parent 229Th (∼7340 y), for radium, were added before evaporation to dryness using a heating lamp, and uranium and radium separation was performed.
Uranium was separated using extraction chromatography on a UTEVA column similar to the total dissolution, while the next section describes the method of radium separation.
Radiochemical separation of radium was modified from the described method15 using 150 mBq 225Ra, in equilibrium with the parent 229Th, as a radiotracer. Care was taken to ensure quantitative transfers until the point of Ra separation, which was achieved by adding 50.0 ml 0.1 M HNO3 to the treated fraction from the sequential extraction, then coprecipitating Ra with PbSO4 by adding consecutively 1.0 ml of concentrated H2SO4, 2.0 g K2SO4 and 1.0 ml of 0.24 M of Pb(NO3)2. The solid was centrifuged in a 50.0 ml tube at 3000 rpm (∼6200 g) for 10.0 minutes, and then washed with 20.0 ml of a mixture of 0.2 M H2SO4/0.1 M K2SO4.
The precipitate was dissolved in 5.0 ml of 0.1 M ethylenediaminetetraacetic acid (EDTA)/NH4OH (pH 10), passed through an anion exchange column (Bio-Rad AG1-X8, 100–200 mesh, chloride form, 5 × 0.5 cm) to remove sulphate and washed with 13.0 ml 0.01 M EDTA/NH4OH. To the eluate Th, 1.0 ml 5.0 M CH3COONH4 was added (pH 4.5) and the solution was passed through a cation exchange column (Bio-Rad AG50W-X12, 200–400 mesh, 8.0 × 0.7 cm) at a flow rate of 1.0 ml minute−1. The column was previously conditioned with 15.0 ml 1.5 M CH3COONH4 followed by 15.0 ml 0.25 M CH3COONH4. Another 50.0 ml 1.5 M CH3COONH4/0.1 M HNO3 was passed through this column to remove Pb and Ac, while Ba was eluted by washing the column with 40.0 ml 2.5 M HCl. Finally, Ra was eluted with 25.0 ml 6.0 M HNO3, and this solution was evaporated to dryness using a heating lamp.
The electrolysis cell consists of two glass tubes (Sovril SV 30) joined with a SV 30 plastic joint. A polished stainless steel planchette (cathode) was held between the two glass tubes by a recessed brass planchette mount supported by the lower electrode. The cell was sealed with a Teflon ring and checked for leaking. A platinum wire anode, inserted in a narrow glass tube, was passed through a rubber bung into the electrolyte solution to complete the electric circuit.
For radium electroplating, the Ra fraction was re-dissolved in organic electrolyte solution (1.0 ml 0.1 M HNO3 in 9.0 ml ethanol) and electroplated on to a stainless steel planchette at 120 mA for 30 minutes. One minute before the end, 1.0 ml of ammonia (s.g. 0.88) was added and the power was turned off. The solution was discarded and the planchette was dried on a hotplate at 200 °C for 5 minutes.
2.7 Stable element analysis
The leachates from sequential extraction of the sediments with the highest uranium content, S3 and S7, were analysed using inductively coupled plasma optical emission spectroscopy (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS), the latter applied where the concentrations were low enough. For analysis of the sediment samples, 1.0 ml of each fraction obtained from the sequential extraction was made up to 10.0 ml with 2% HNO3. The samples were run using a series of solutions prepared from certified standard solutions (1000 ppm, Sigma Aldrich, UK) for each analyte.2.8 Sediment characterisation
The instrument is equipped with five wavelength detectors and it was also possible to use these to obtain elemental maps using wavelength-dispersive spectroscopy after using suitable standard materials. The 10 elements selected were divided into two groups. The first group included U, Ca, Mg, Mn and Fe, while K, Cu, As, Sn and Pb were in the second group.
3. Results and discussion
3.1 Physico-chemical properties of sediments
The bulk minerals identified by X-ray diffraction (XRD) and the organic matter (OM) content calculated from the loss of ignition are outlined in ESI (Table S2†). The OM content of the River Fal sediment is much lower (1.0%) than that of the sediments collected from streams running towards the river. In some streams, the OM content of the sediments is as high as 37% (S3), 25% (S2) and 21% (S7). Organic-rich sediments are also observed in some streams emerging from adits (e.g. 29% in S12 and 20% in S13) along the river course.XRD is only useful in characterising crystalline minerals, so minerals with disordered structures will not give well defined patterns. The mineralogical composition of the bulk samples reveals that quartz is the main component of all sediments, as expected in stream sediments. In addition muscovite, kaolinite, rutile and schorl are identified in all samples. It is important to note that the lowest percentage of quartz was found in the sample with the highest uranium content (S7), and the highest proportion of phyllosilicate minerals (muscovite and kaolinite). Minor dolomite and chlorite were also identified, addition to jarosite (found in one sample coming from an adit). Overall though, the results from XRD suggest no substantive difference in bulk sediment composition, whether for those collected from the main river or from the side streams.
3.2 Radiochemical characterisation of sediments
The specific activities and activity ratios of the natural radionuclides 238U, 234U and 226Ra in the sediment from the River Fal and streams around South Terras mine are presented in Table 2.| ID | 238U | 234U | 226Ra | 234U/238U | 226Ra/238U | Latitude (N) | Longitude (W) |
|---|---|---|---|---|---|---|---|
| S1 | 230 ± 12 | 181 ± 10 | 293 ± 15 | 0.79 ± 0.06 | 1.27 ± 0.09 | 50° 19.861′ | 4° 54.248′ |
| S2 | 290 ± 14 | 260 ± 13 | 424 ± 23 | 0.90 ± 0.06 | 1.46 ± 0.10 | 50° 19.863′ | 4° 54.257′ |
| S3 | 1820 ± 36 | 1388 ± 32 | 940 ± 53 | 0.76 ± 0.02 | 0.52 ± 0.03 | 50° 19.856′ | 4° 54.248′ |
| S4 | 43 ± 5 | 44 ± 5 | 55 ± 6 | 1.02 ± 0.18 | 1.28 ± 0.20 | 50° 19.809′ | 4° 54.274′ |
| S5 | 49 ± 6 | 55 ± 6 | 68 ± 8 | 1.12 ± 0.19 | 1.39 ± 0.23 | 50° 19.810′ | 4° 54.272′ |
| S6 | 160 ± 10 | 127 ± 9 | 220 ± 12 | 0.79 ± 0.08 | 1.38 ± 0.12 | 50° 19.809′ | 4° 54.275′ |
| S7 | 4350 ± 53 | 4265 ± 52 | 1765 ± 48 | 0.98 ± 0.02 | 0.41 ± 0.01 | 50° 20.014′ | 4° 54.330′ |
| S8 | 95 ± 8 | 104 ± 8 | 116 ± 11 | 1.09 ± 0.13 | 1.22 ± 0.15 | 50° 20.059′ | 4° 54.344′ |
| S9 | 43 ± 5 | 39 ± 5 | 75 ± 7 | 0.91 ± 0.16 | 1.74 ± 0.26 | 50° 20.103′ | 4° 54.321′ |
| S10 | 51 ± 5 | 57 ± 5 | 61 ± 5 | 1.12 ± 0.14 | 1.20 ± 0.15 | 50° 19.559′ | 4° 54.259′ |
| S11 | 40 ± 6 | 40 ± 6 | 61 ± 5 | 1.00 ± 0.19 | 1.53 ±![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.24 0.24 |
50° 19.559′ | 4° 54.260′ |
| S12 | 39 ± 5 | 35 ± 5 | 194 ± 16 | 0.90 ± 0.17 | 4.97 ±0.75 |
50° 19.558′ | 4° 54.260′ |
| S13 | 44 ± 5 | 54 ± 6 | 72 ± 7 | 1.23 ± 0.20 | 1.64 ± 0.26 | 50° 19.547′ | 4° 54.233′ |
| S14 | 50 ± 6 | 49 ± 6 | 67 ± 6 | 0.98 ± 0.16 | 1.34 ±0.19 |
50° 19.550′ | 4° 54.233′ |
| S15 | 49 ± 6 | 52 ± 6 | 60 ± 5 | 1.06 ± 0.18 | 1.22 ±0.18 |
50° 19.014′ | 4° 54.330′ |
| S16 | 44 ± 6 | 42 ± 6 | 53 ± 4 | 0.95 ± 0.18 | 1.20 ± 0.19 | 50° 19.359′ | 4° 54.019′ |
| S17 | 13 ± 3 | 16 ± 4 | 42 ± 4 | 1.23 ± 0.39 | 3.23 ± 0.80 | 50° 19.371′ | 4° 54.010′ |
| S18 | 43 ± 6 | 42 ± 6 | 62 ± 5 | 0.98 ± 0.18 | 1.44 ±0.21 |
50° 19.422′ | 4° 54.097′ |
| S19 | 41 ± 5 | 40 ± 5 | 53 ± 4 | 0.98 ± 0.19 | 1.29 ±0.22 |
50° 19.701′ | 4° 54.286′ |
| S20 | 73 ± 7 | 66 ± 6 | 51 ± 4 | 0.90 ± 0.12 | 0.70 ± 0.09 | 50° 20.138′ | 4° 54.352′ |
The sample located about 100 m upstream (S20) was selected to provide a background level for the sediments in the River Fal. This point shows negligible influence from the mine on the uranium activity (73 Bq kg−1), while the concentration of radium is 51 Bq kg−1. Moreover, the ratios of 234U/238U and 226Ra/238U in S20 are close to those observed far from uranium mines.16 The highest concentrations of the radionuclides are found in the sediments collected downstream, about 135 m south of the mine building (S7). The values are up to 4350, 4265 and 1765 Bq kg−1 for 238U, 234U and 226Ra, respectively. The ratio between the two uranium isotopes, within 1σ, is around one, indicating equilibrium. This is probably because the parent materials of these sediments released from the old mine heaps and are relatively new, which initially had 234U/238U activity ratio near 1.0 and has not been subjected to much leaching of 234U since its deposition.
At a distance of about 425 m downstream, the concentrations of the radionuclides vary in the small streams draining towards the River Fal. For samples S1 and S2, the specific activities of 238U are 230 and 290 Bq kg−1. These samples were depleted slightly in the daughter, 234U, compared with the parent, 238U. However, within the same area, sample S3 displayed 238U content of 1820 Bq kg−1, with a similar depletion in 234U as S1 and S2. In addition, the 226Ra shows a specific activity higher than the background in the sediments from these streams, with a value of 940 Bq kg−1 at S3.
For the remainder of the samples, with the exception of the 226Ra level in S6 and S12, the sediments in the River Fal seem to display local background levels (range 42 to 115 Bq kg−1 with mean value 64 Bq kg−1). Since sediments can indicate the source of contamination in an area, the results from the River Fal suggest that the South Terras mine has no significant radiological impact on the nearby water courses beyond a distance greater than 0.5 km from the mine buildings.
3.3 Sequential chemical extraction results
Elevated specific activities of 238U, 234U and 226Ra were observed in the two samples (S3 and S7) collected close to the mine site. In order to identify the association of the radionuclides with the different geochemical fractions, sequential chemical extraction (SCE) was performed.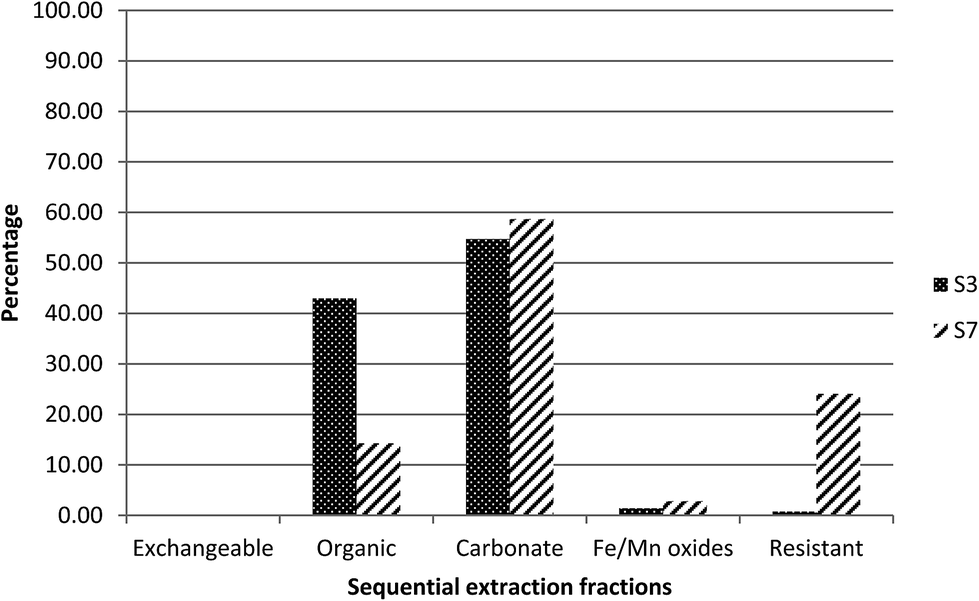 | ||
| Fig. 2 Extraction profile of uranium as a percentage of the sum of five fractions in S3 and S7. | ||
For radium, as can be seen in Fig. 3, about 70% was found to be associated with fraction 2, the organic fraction, while about 15% was released in fraction 3, the carbonate fraction, and only 10% was released in fraction 1, the exchangeable fraction. This may be linked to the higher organic matter (37%) in this sediment.19
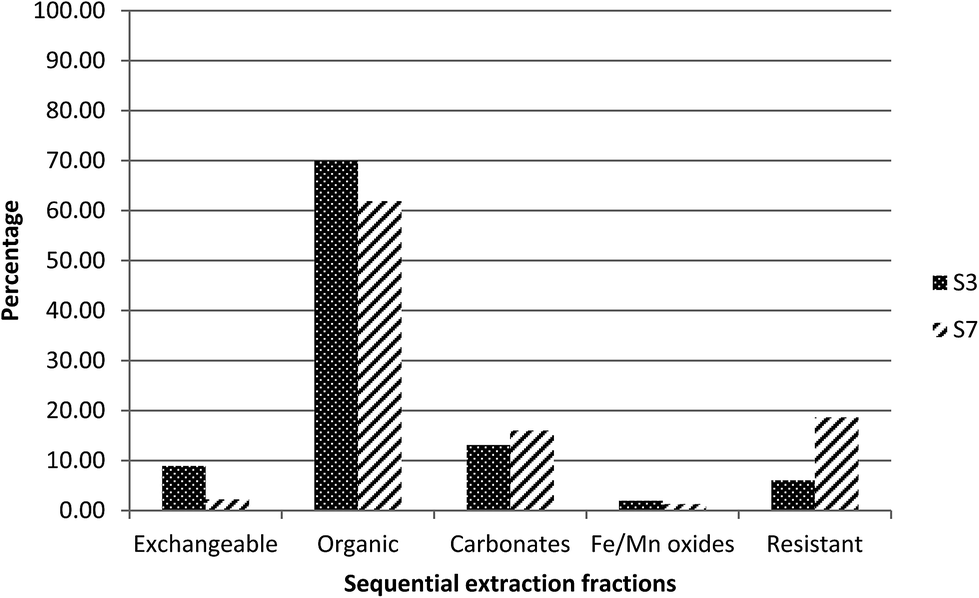 | ||
| Fig. 3 Extraction profile of radium as a percentage of the sum of five fractions in S3 and S7. | ||
This is again consistent with the proposed association between uranium and carbonate phases. However, compared to sample S3, a higher percentage of uranium was present in the residual fraction, fraction 5, suggesting the presence of primary U-minerals in S7.
For radium, 60% was found to be associated with fraction 2, the organic fraction, and 20% was released in fraction 5, the residual fraction (Fig. 3). In contrast to S3, part of the radium appears to be associated with a resistant phase, rather than primarily controlled by adsorption. This would be reasonable, given the presence of uranium in the residual fraction and therefore the potential for in situ generation of radium.
3.4 Radionuclide and stable element fractionation
Comparing the geochemical distribution of the stable elements with the radionuclides in S3 and S7 could help understanding of distribution between the operationally-defined phases. Accordingly, geochemical fractionation of selected stable elements was undertaken in order to explore the most likely geochemical host phases for uranium and radium in Cornwall sediments.Calcium was associated mainly with the fractions 1 and 2 (exchangeable and organic) fractions in both samples (ESI Fig. S1†), with the majority of the calcium (∼80%) in S7 bound to fraction 2 compared with only 60% in S3.
Manganese was also associated primarily with fractions 1 and 2, the exchangeable and organic matter fractions, in S3, with nearly the same percentage in each fraction (ESI Fig. S2†). However, in S7 it was found in all fractions, with a relatively higher percentage (∼40%) in fraction 2 (organic).
Fractionation of titanium and iron are illustrated in ESI (Fig. S3 and S4†). In both samples, the majority of titanium (∼90%) was released from fraction 5, the fraction leached by strong acids. This is consistent with the classification of Ti as a refractory element,20 which, therefore, is expected to be bound to the resistant fraction. In both sediments, iron, same as titanium, was also found mainly in this fraction, with about 75%.
Arsenic was found to associate with fractions 2 and 3 of S7, with ∼60% in carbonate and ∼37% in organic (ESI Fig. S5†). In S3, it was more distributed towards the acid-resistant fractions, with about 30% in fraction 5, the resistant component.
Barium fractionation is very similar to that of Ra (ESI Fig. S6†). In S3, it was attached predominantly to fraction 2; however, for S7 it was distributed in all fractions, with the exception of fraction 4 (Fe/Mn oxides), with a relatively higher amount (∼40%) in fraction 5, the resistant fraction.
Comparing the geochemical fractionation of stable elements with that of uranium and radium showed that, in both S3 and S7, the uranium distribution was quite different from that of any stable elements. In both sediments, the fractionation profile of barium showed some similarities to that of radium, as expected from the similarity in their chemistries.
3.5 Uranium isotopic ratios in sequential extraction fractions
The activity ratio between 238U and 234U isotopes was used to obtain information about the mobility of uranium between water-solid phases. This is because 234U is expected to be more abundant in water relative to 238U, and the vice versa is expected in sediment. The uranium isotopic activity ratio (234U/238U) in the sequential extraction fractions of samples S3 and S7, excluding the exchangeable fraction, is shown in Fig. 4 and 5.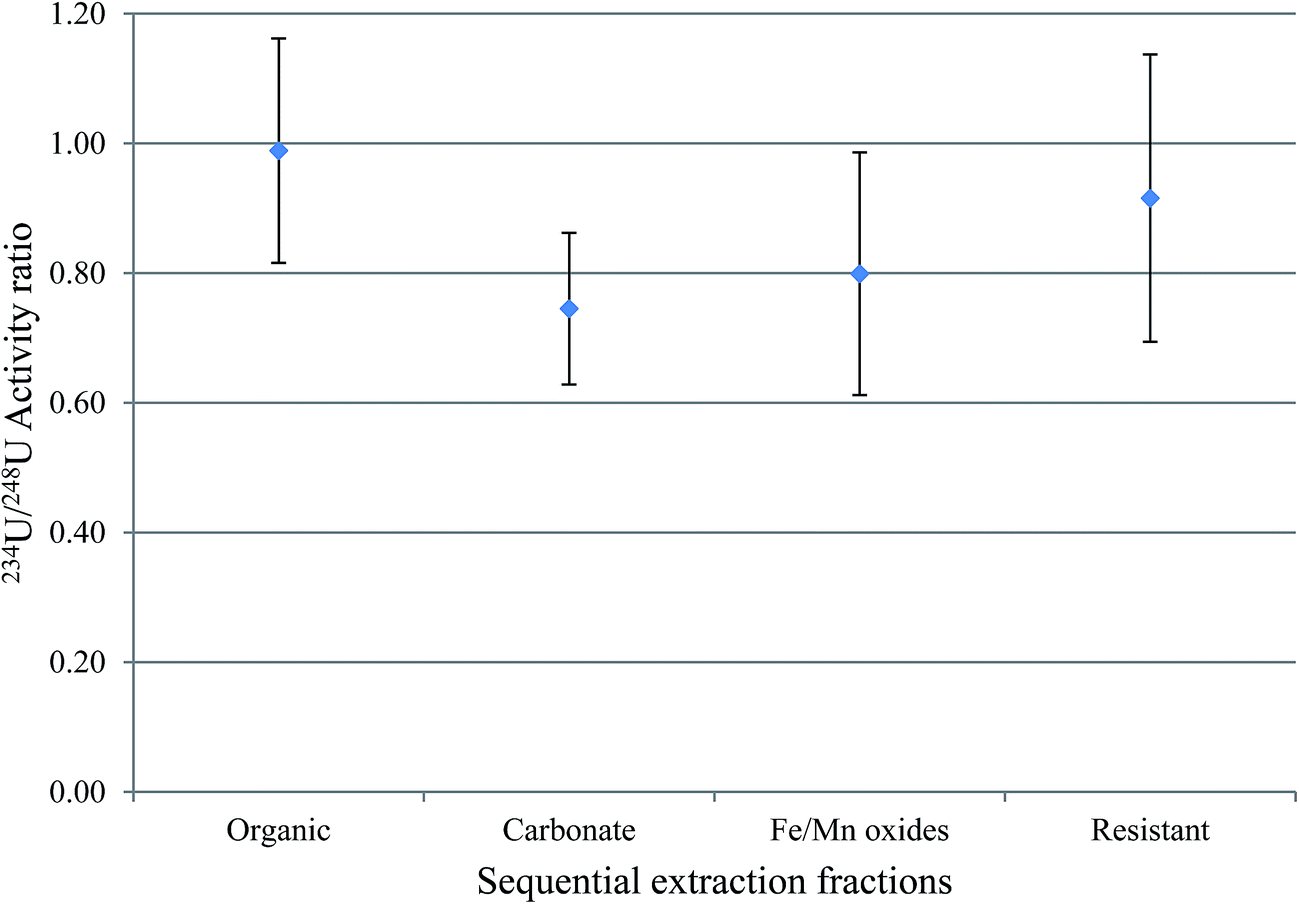 | ||
| Fig. 4 234U/238U activity ratios in the sequential extraction fractions of S3. | ||
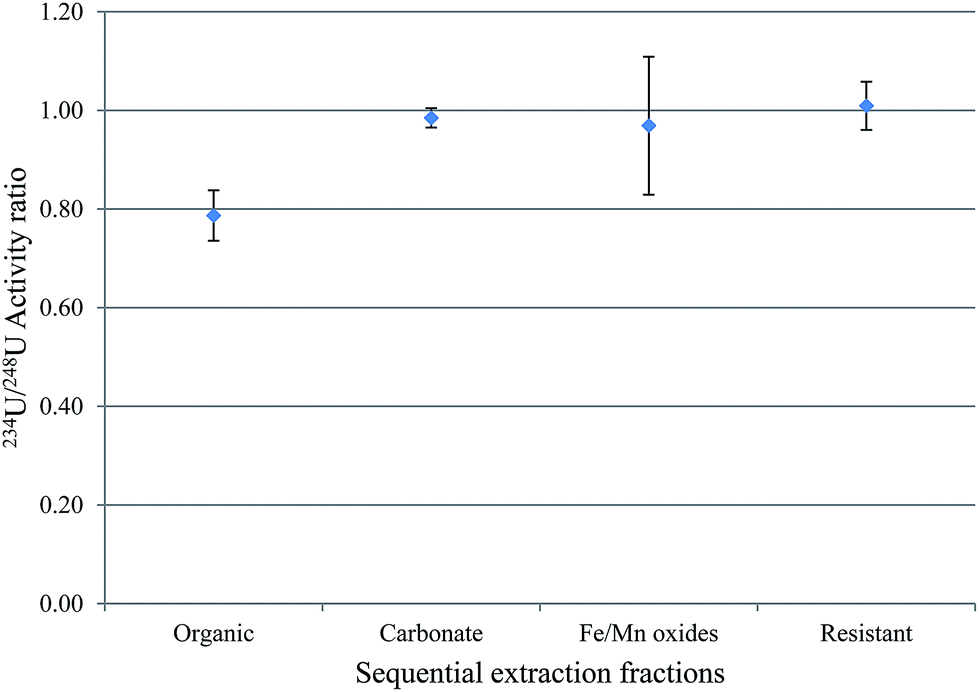 | ||
| Fig. 5 234U/238U activity ratios in the sequential extraction fractions of S7. | ||
For S3, the results indicated that, within 1σ, the 234U/238U activity ratios in fractions 2 (the organic fraction), 4 (Fe/Mn oxides) and 5 (the resistant) were within equilibrium, with the organic the closest to unity; however, for the carbonate fraction, uranium isotopic ratios was clearly lower than unity. As mentioned above, S3 has the highest OM content of all these sediments (∼40%) and OM plays an important role in retaining radionuclides. A possible reason for this higher 234U/238U in the organic fraction compared with the other fractions is that the organic fraction, which will include organic coatings on other particles, is more likely to be in contact with water, and therefore has a great opportunity to adsorb uranium from water, with an activity ratio greater than unity.21
For S7, fractions 3, 4 and 5, the carbonates, Fe/Mn oxides and the resistant, within 1σ, revealed 234U/238U activity ratios within equilibrium. However, the 234U/238U activity ratio in fraction 2, the organic, was ca. 0.8, indicating substantial disequilibrium. As discussed earlier, disequilibrium in sediments is interpreted as relating to the preferential leaching of 234U from the mineral grain, compared with the parent 238U. Consequently, water is expected to exhibit 234U/238U > 1, while sediment is expected to show 234U/238U < 1.22–24 In S7, which has the highest uranium content of these sediments, the equilibrium 234U/238U ratio may reflect the presence of significant primary U minerals (hence the relatively high proportion of uranium in fraction 5, the resistant component), and this material could contribute uranium to fractions 3 and 4, giving the same ratio. By contrast, the much lower ratio in fraction 2 suggests a more complex process, for example, may be, derivation of the uranium in this fraction from another source.25
3.6 Characterisation of sediments using spectroscopic methods
 | ||
| Fig. 6 Scanning electron microscope (SEM) results showing backscattered electron (BSE) images (A) and electron-dispersive spectroscopy (EXD) spectrum (B) of the bulk minerals of S3. The bright area is an indication of a presence of high atomic number elements. | ||
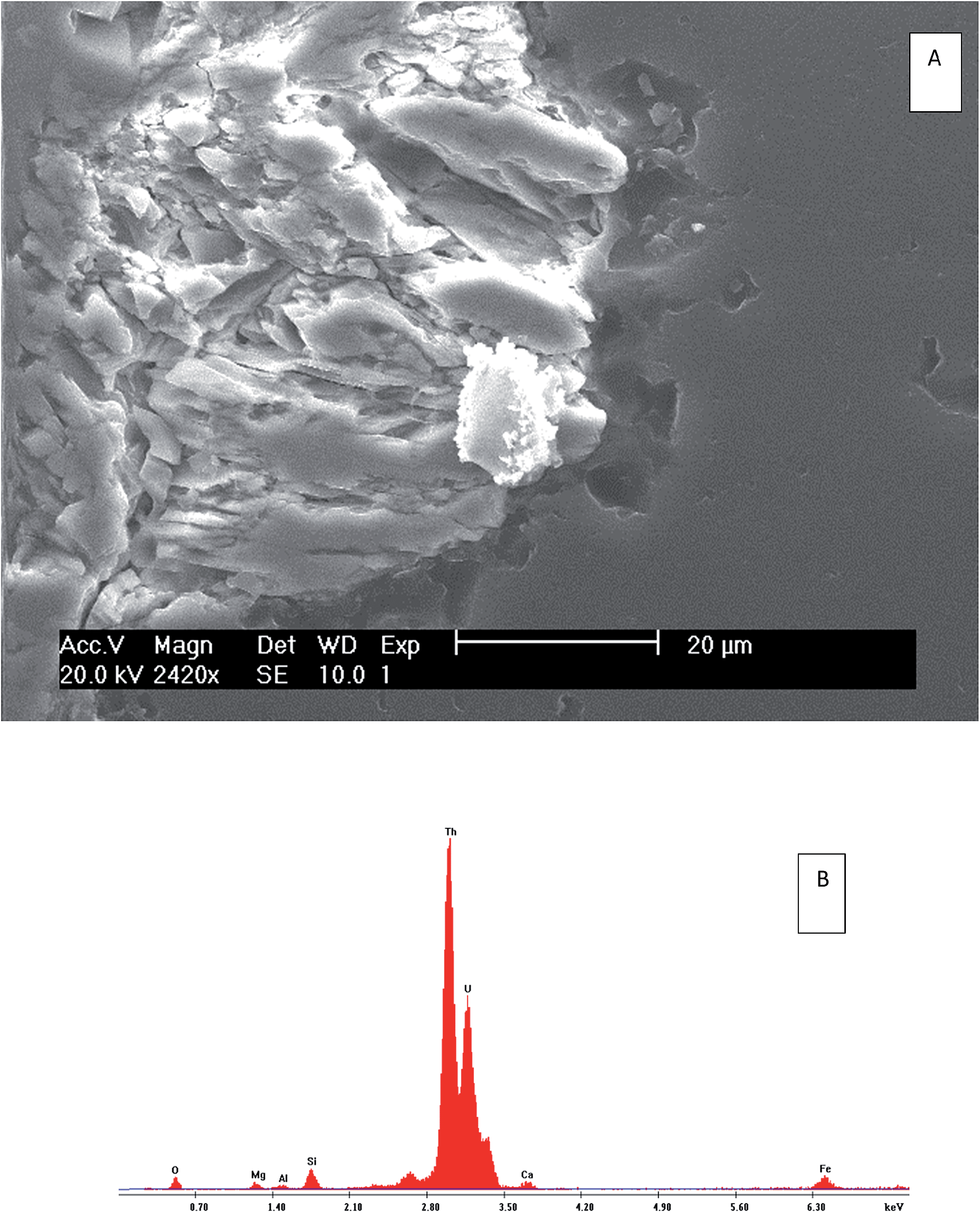 | ||
| Fig. 7 Scanning electron microscope (SEM) results showing secondary electron (SE) images (A) and electron-dispersive spectroscopy (EXD) spectrum (B) of the bulk minerals of S3. | ||
The SEM/EDS results of the heavy minerals of S7, isolated using heavy liquid separation, revealed the presence of U-bearing particles, as presented in Fig. 8 and 9. The EDS spectrum (Fig. 8) identified an association of uranium with P, Ca and Th in one particle. The percentage of the oxides forms were (9.83%, 0.21%, 0.22%) respectively, compared with 0.18% of uranium. In another particle (Fig. 9), the uranium was found to associate with clay minerals (Al and Si). This may support the sequential extraction results, where a considerable amount (25%) of uranium in S7 was released from the residual fraction, while, for S3, only 1% of the uranium was released from same fractions.
 | ||
| Fig. 8 Scanning electron microscope (SEM) results showing secondary electron (SE) images (A) and electron-dispersive spectroscopy (EXD) spectrum (B) of a single grain isolated from the heavy minerals separated by heavy liquid from the richest U-sample (S7). A U association with P, Th and Ca (from the EDX analysis) has been identified. | ||
 | ||
| Fig. 9 Scanning electron microscope (SEM) results showing secondary electron (SE) images (A) and electron-dispersive spectroscopy (EXD) spectrum (B) of the heavy minerals separated by heavy liquid fractionation of S7. A U association with aluminosilicates has been identified. | ||
 | ||
| Fig. 10 Backscattered electron (BSE) image (top) and X-ray maps of elements (Mn, K, Fe, As, Ca and U) from electron microprobe analysis (EMPA) of the heavy mineral fraction from S7. | ||
For S3, EMPA characterisation of the bulk minerals could not identify uranium-rich grains. However, for S7, a grain from the heavy minerals separated by the heavy liquid was identified as containing trace amounts of uranium-bearing minerals (Fig. 10). The WDS images suggested that the grain includes a higher amount of K, Ca and Fe relative to U, As and Mn. The percentage of uranium oxide (wt% UO2) in this grain was about 1%.
4. Conclusions
Radioactivity around the former uranium mining site at South Terras is generally close to local background levels, with no substantial effect of the radionuclides on the River Fal, and enhanced concentrations of radionuclides only found in the immediate area of the mine. The elevated activity at distances less than 0.5 km could be related to the migration of particles enriched in uranium from the mine location due to weathering.Sequential chemical extraction, applied to the sediments with the highest radionuclide concentrations, revealed different geochemical fractionation of uranium and radium. The uranium in the sediment with the highest organic matter was more closely associated with relatively labile fractions, particularly the organic and the carbonate fractions. Furthermore, the sample with the highest uranium revealed that although the carbonate bound more of the uranium, a significant portion was held in the resistant fraction. Radium in both sediments was held primarily in the organic fraction but, in S7, the sample with the highest radioactivity, significant radium was also held in the resistant fraction.
There was no clear association of the radionuclides and the stable elements in individual fractions, although there were indications of an association with calcium, manganese and arsenic in the organic and the carbonate fractions.
The activity ratios of the uranium isotopes may suggest that exchange between sediment/water could explain the findings so further study, including measurements of the uranium isotopic ratios in water samples, is recommended.
Using SEM, uranium-bearing particles have been localised in the bulk minerals and the heavy minerals from the sediments enriched in uranium. EMPA results produced an X-ray map of the uranium, with associated stable elements, in a single grain obtained from the sample with the highest uranium content.
Acknowledgements
The authors greatly appreciate financial support from the Islamic Development Bank (IDB), Jeddah, Saudi Arabia. The authors thank Mr Paul Lythgoe for ICP-OES/ICP-MS measurements, Mr Alastair Bewsher for IC analysis, Mrs Cath Davies for total dissolution of samples, Dr John Waters for SEM and Dr John Charnock for EMPA analysis, all from School of Earth, Atmospheric and Environmental Sciences, University of Manchester. The authors are grateful to the Boconnoc Estate for permission to access the South Terras mine site, and to local landowners for permission to access other locations.References
- A. J. Plater, M. Ivanovich and R. E. Dugdale, Appl. Geochem., 1992, 7, 101–110 CrossRef CAS.
- F. P. Carvalho, J. M. Oliveira, I. Lopes and A. Batista, J. Environ. Radioact., 2007, 98, 298–314 CrossRef CAS PubMed.
- B. S. Marko Strok, J. Environ. Radioact., 2010, 101, 22–28 CrossRef PubMed.
- F. P. Carvalho and J. M. Oliveira, J. Radioanal. Nucl. Chem., 2007, 274, 167–174 CrossRef CAS.
- G. R. Hancock, M. K. Grabham, P. Martin, K. G. Evans and A. Bollhofer, Sci. Total Environ., 2006, 354, 103–119 CrossRef CAS PubMed.
- P. Blanco, F. V. Tome and J. C. Lozano, J. Environ. Radioact., 2005, 79, 315–330 CrossRef CAS PubMed.
- G. K. Gillmore, P. S. Phillips, G. Pearce and A. Denman, International radon symposium, 2001, pp. 94–105 Search PubMed.
- Y. Moliner-Martinez, P. Campins-Falco, P. J. Worsfold and M. J. Keith-Roach, J. Environ. Monit., 2004, 6, 907–913 RSC.
- O. W. Purvis, E. H. Bailey, J. McLean, T. Kasama and B. J. Williamson, Geomicrobiol. J., 2004, 21, 159–167 CrossRef CAS.
- R. A. Sutherland, Hydrobiologia, 1998, 389, 153–167 CrossRef CAS.
- G. G. Kipp, J. J. Stone and L. D. Stetler, Appl. Geochem., 2009, 24, 2246–2255 CrossRef CAS PubMed.
- H. E. Carter, P. Warwick, J. Cobb and G. Longworth, Analyst, 1999, 124, 271–274 RSC.
- I. Eichrom Technologies 2001.
- M. K. Schultz, K. G. W. Inn, Z. C. Lin, W. C. Burnett, G. Smith, S. R. Biegalski and J. Filliben, Appl. Radiat. Isot., 1998, 49, 1289–1293 CrossRef CAS.
- K. A. Smith and E. R. Mercer, J. Radioanal. Chem., 1970, 5, 303–312 CrossRef CAS.
- J. C. Lozano, P. Blanco Rodríguez and F. Vera Tomé, J. Environ. Radioact., 2002, 63, 153–171 CrossRef CAS.
- N. G. Rachkova, I. I. Shuktomova and A. I. Taskaev, Eurasian Soil Sci., 2010, 43, 651–658 CrossRef.
- G. G. Kipp, J. J. Stone and L. D. Stetler, Appl. Geochem., 2009, 24, 2246–2255 CrossRef CAS PubMed.
- D. J. Greeman, A. W. Rose, J. W. Washington, R. R. Dobos and E. J. Ciolkosz, Appl. Geochem., 1999, 14, 365–385 CrossRef CAS.
- M. K. Schultz, W. Burnett, K. G. W. Inn and G. Smith, J. Radioanal. Nucl. Chem., 1998, 234, 251–256 CrossRef CAS.
- M. J. Vargas, F. V. Tome, A. M. Sanchez, M. T. C. Vazquez and J. L. G. Murillo, Appl. Radiat. Isot., 1997, 48, 1137–1143 CrossRef CAS.
- N. Vigier, B. Bourdon, E. Lewin, B. Dupre, S. Turner, G. J. Chakrapani, P. van Calsteren and C. J. Allegre, Chem. Geol., 2005, 219, 69–91 CrossRef CAS PubMed.
- N. Vigier, K. W. Burton, S. R. Gislason, N. W. Rogers, S. Duchene, L. Thomas, E. Hodge and B. Schaefer, Earth Planet. Sci. Lett., 2006, 249, 258–273 CrossRef CAS PubMed.
- J. Riotte and F. Chabaux, Geochim. Cosmochim. Acta, 1999, 63, 1263–1275 CrossRef CAS.
- S. M. Siddeeg, N. D. Bryan and F. R. Livens, Environ. Sci.: Processes Impacts, 2014, 16, 991–1000 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4em00230j |
| This journal is © The Royal Society of Chemistry 2015 |