
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2,2′-Disilylazobenzenes featuring double intramolecular nitrogen⋯silicon coordination: a photoisomerizable fluorophore†
Naokazu
Kano
*,
Masaki
Yamamura
and
Takayuki
Kawashima‡
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kano@chem.s.u-tokyo.ac.jp
First published on 24th July 2015
Abstract
(E)-4,4′-Dimethyl-2,2′-disilylazobenzenes were synthesized. Double intramolecular N⋯Si coordination in the bis(fluorodimethylsilyl) and bis(trifluorosilyl) derivatives was confirmed using X-ray crystallographic analysis and 29Si NMR spectroscopy. In the absorption bands, due to the π,π* transitions, introduction of silyl groups was found to cause a bathochromic shift. In contrast to most azobenzenes, which do not fluoresce at all, the (E)-2,2′-bis(trifluorosilyl)azobenzene derivative with the N⋯Si coordination fluoresced a yellow-green colour at room temperature. Methyl and trifluorosilyl groups lowered the n and π* orbitals, as revealed by DFT calculations. As a result, the lowest singlet excitation energy state is found to be the allowed π,π* transition, different from the forbidden n,π* transition in general azobenzenes, as revealed by TD-DFT calculations. The allowed transition character of the lowest singlet excited state and moderately rigid conformation of the azo moiety, provided by the double N⋯Si coordination, account for the fluorescence emission. Nevertheless, the N⋯Si coordination is weak enough to be cleaved upon photoexcitation, and thus the (E)-2,2′-disilylazobenzenes undergo photoisomerization to the (Z)-isomers. Both the photoisomerization and fluorescence emission properties of the azobenzene moiety have been achieved for the first time. After photoisomerization of the (E)-2,2′-disilylazobenzenes to the corresponding (Z)-isomer, they do not fluoresce. This change in the fluorescence intensity upon photoisomerization is useful for the regulation of fluorescence properties. Therefore, this compound can be recognized as a unique photoisomerizable fluorophore to regulate the fluorescence intensity using a single light source.
Introduction
Azobenzene, which is the fundamental core of azo dyes, is popular because of its reversible isomerization between the (E)- and (Z)-isomers upon photoirradiation. The photoisomerization properties of azobenzene have been applied to molecular switches, photoregulation of biomaterials, light-driven molecular machines and molecular logic gates.1 Its photoisomerization mechanism has been a subject of interest in photochemistry.2 The photoisomerization properties of azobenzene impede its fluorescence emission, which is another property of some dyes.3 Azobenzene derivatives are usually non-fluorescent or virtually non-fluorescent. Only a few azobenzene derivatives fluoresce as a fluorophore as a result of freezing in a matrix at low temperature,4ortho-metalation,5 liquid crystal formation6 or self-assembled aggregation.7 In addition, whereas there are several photochromic switches for modulation of fluorescence emission,8 there are only a few examples of azobenzene derivatives that undergo both photoisomerization and measurable fluorescence emission from their azobenzene moieties.9 Such a photoisomerizable fluorophore is interesting for the purpose of regulation of fluorescence intensity by a single light source.10We have previously achieved intense fluorescence emission from (E)-2-borylazobenzenes (E)-1 containing an electron-withdrawing bis(pentafluorophenyl)boryl group near the azo group (Chart 1).11 A key factor for the fluorescent emission was the existence of an intramolecular nitrogen–boron interaction that could change the order of the singlet excited state by lowering the energy level of the n orbital of the azo group and locking the conformation to inhibit the isomerization. In (E)-2,2′-diborylazobenzenes (E)-2, the twofold intramolecular N–B interactions greatly lowered the energy level of the n orbital, facilitating its single-electron reduction.12 The strong N–B interactions, however, suppressed photoisomerization, which is the most common property of azobenzene, in both (E)-1 and (E)-2. Thus, the fluorescent emission property was found to be gained at the cost of the photoisomerization property.
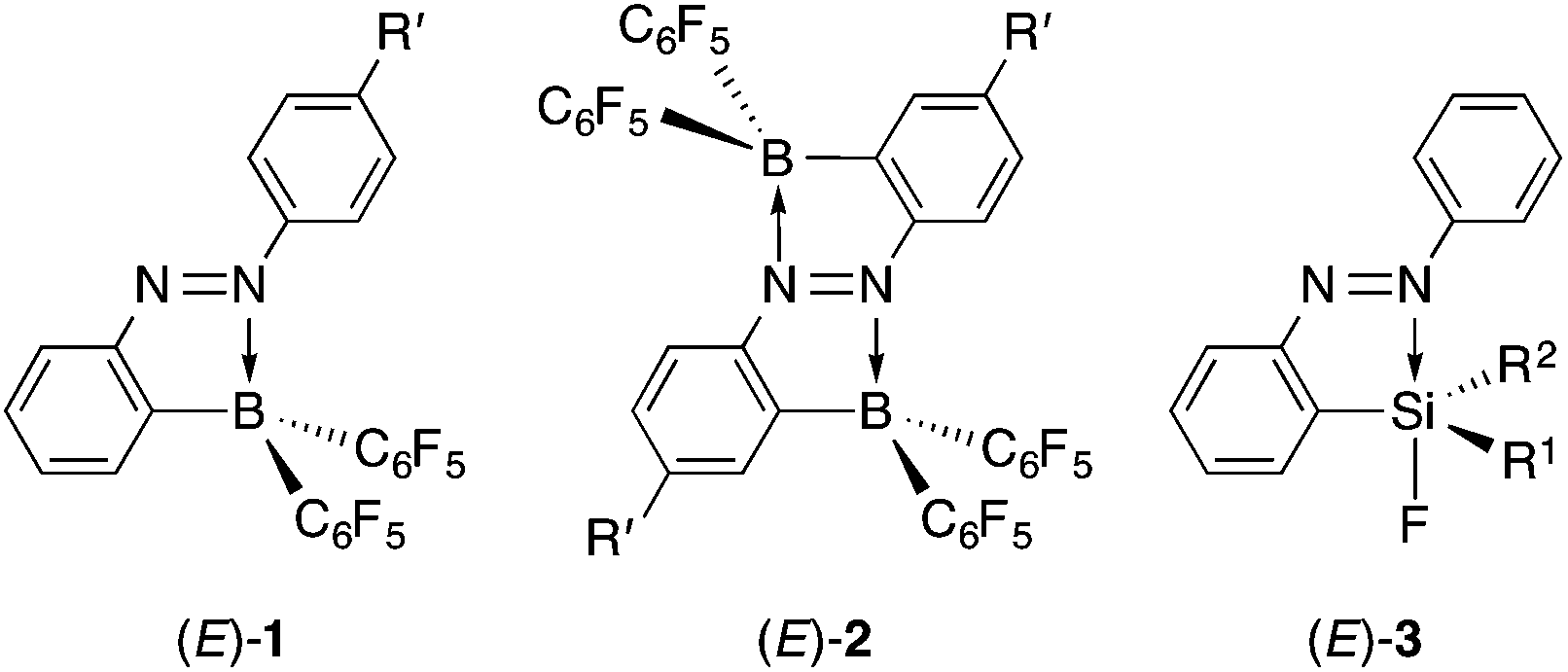 | ||
| Chart 1 | ||
In our previous reports, we have found that several (E)-2-silylazobenzenes (E)-3, which bear an intramolecular nitrogen⋯silicon interaction, do not fluoresce.13 Compounds (E)-3 undergo photoisomerization accompanied by dissociation of the interaction in moderate to almost quantitative yields, indicating the weakness of the nitrogen⋯silicon interaction compared to the nitrogen–boron interaction. (E)-2,2′-Disilylazobenzenes featuring twofold weak nitrogen⋯silicon interactions are promising candidates for an isomerizable fluorophore because a few π-conjugated compounds containing the nitrogen⋯silicon interaction have been recently reported to fluoresce.14 Here, we report the synthesis and structures in both the crystalline and solution states of (E)-2,2′-disilylazobenzenes featuring intramolecular nitrogen⋯silicon interactions, together with their photophysical properties including photoisomerization and fluorescence emission. Their optical properties are discussed based on their spectroscopic measurements and DFT calculations.
Results and discussion
Synthesis
(E)-4,4′-Dimethyl-2,2′-disilylazobenzenes (E)-4a–c (Chart 2) were synthesized from (E)-2,2′-diiodo-4,4′-dimethylazobenzene and the corresponding chlorosilanes via the dilithiated (E)-4,4′-dimethylazobenzene according to the synthetic procedure for (E)-2,2′-bis(diphenylsilyl)-4,4′-dimethylazobenzene.15 Methyl groups at the para-positions are necessary for their good solubility. Fluorination of (E)-4b with AgF gave (E)-4d bearing one fluorine atom on each silicon atom. Compound (E)-4e was synthesized by fluorination of (E)-4c with BF3·OEt2. Compounds (E)-4a–e can be handled in air in the solid state, but (E)-4d,e are moisture sensitive and undergo hydrolysis easily in the solution state.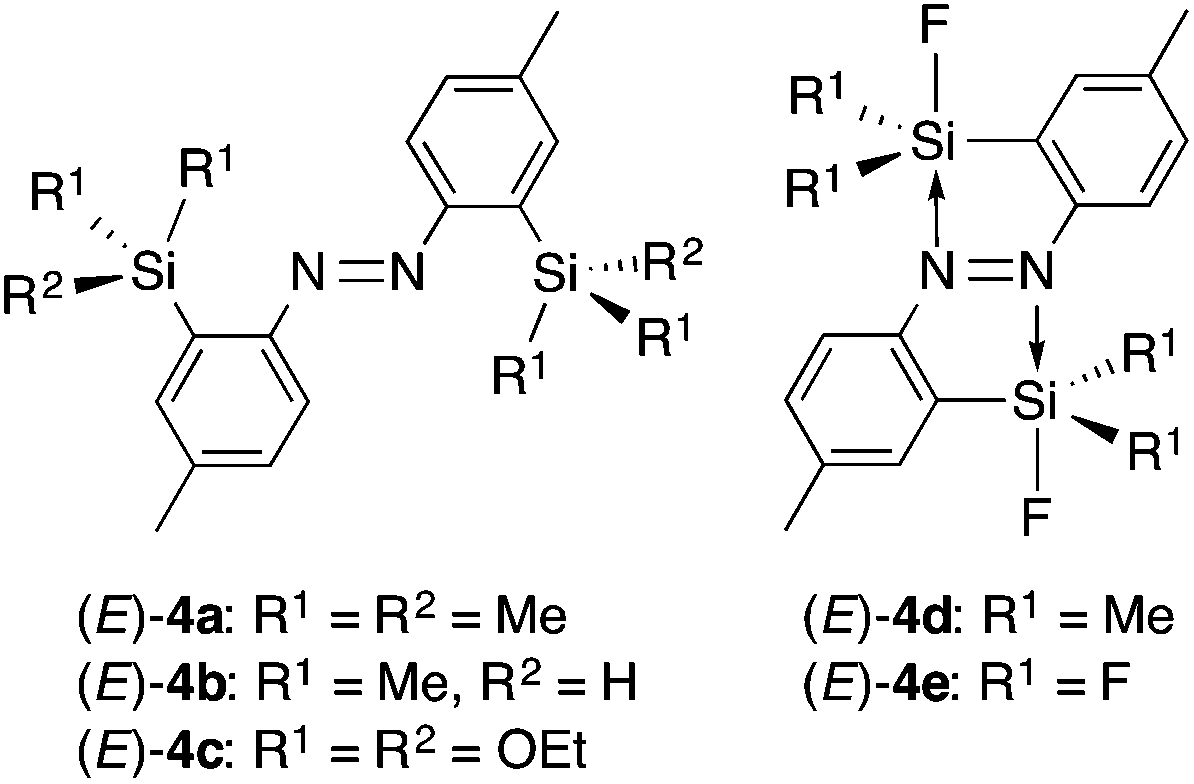 | ||
| Chart 2 | ||
Crystal structure
The structures of (E)-4,4′-dimethyl-2,2′-disilylazobenzenes (E)-4a,d,e were determined using single-crystal X-ray crystallography to reveal the effects of the two silyl groups on the azobenzene structure. Their thermal ellipsoid plots are shown in Fig. 1. Selected bond lengths, bond angles and torsion angles are summarized in Table 1. All of the crystals show an almost planar structure of the azobenzene moiety. In (E)-4a, neither of the two silyl groups in a tetrahedral structure show an interaction with the azo moiety. This configuration is reasonable as a result of steric repulsion between the azo group and trimethylsilyl groups.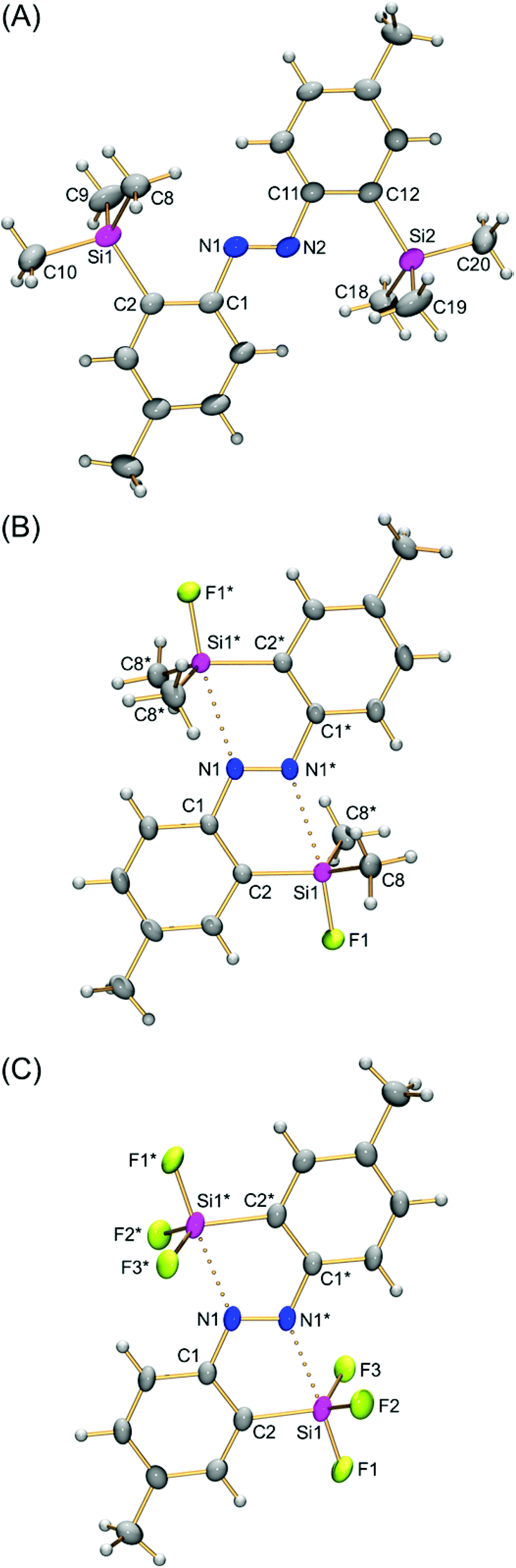 | ||
| Fig. 1 Thermal ellipsoid plots (50% probability) of (E)-2,2′-disilylazobenzenes (E)-4a (A), (E)-4d (B) and (E)-4e (C). Independent molecules of (E)-4a in the crystal unit cell are omitted. | ||
| (E)-4a | |||
| N1–N2 | 1.255(4) | C8–Si1–C10 | 107.4(2) |
| C2–Si1–C8 | 112.77(17) | C9–Si1–C10 | 108.6(2) |
| C2–Si1–C9 | 110.03(19) | C1–N1–N2 | 114.1(3) |
| C2–Si1–C10 | 107.18(18) | C2–C1–N1–N2 | 179.3(3) |
| C8–Si1–C9 | 110.6(2) | ||
| (E)-4d | |||
| N1–N1* | 1.274(4) | C2–Si1–C8 | 115.57(8) |
| N1*⋯Si1 | 2.585(3) | C8–Si1–C8* | 119.09(14) |
| F1–Si1–C2 | 100.27(11) | C1–N1–N1* | 113.4(3) |
| F1–Si1–C8 | 100.63(8) | C2–C1–N1–N1* | 0.0(0) |
| (E)-4e | |||
| N1–N1* | 1.255(6) | F2–Si1–F3 | 110.32(16) |
| N1*⋯Si1 | 2.371(4) | C2–Si1–F2 | 119.95(17) |
| F1–Si1–F2 | 99.93(16) | C2–Si1–F3 | 118.81(16) |
| F1–Si1–F3 | 101.11(16) | C1–N1–N1* | 113.3(4) |
| F1–Si1–C2 | 102.18(17) | C2–C1–N1–N1* | 0.6(5) |
In contrast, both silyl groups in (E)-4d,e have the syn configuration with the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond against the C1–N1 or C1*–N1* bond, as shown by the C2–C1–N1–N1* torsion angle, which is close to or equal to 0°. Nitrogen atoms occupy the posterior positions of a Si1–F1 bond with the N1*⋯Si1 interatomic distance (2.371(4)–2.585(3) Å) within the sum (3.65 Å) of the van der Waals radii of both elements.16 The F1–Si1–Cn and F1–Si1–Fn′ bond angles are around 100° and narrower than the bond angle for a tetrahedral structure. These structural parameters show a capped tetrahedral coordination sphere at the silicon atoms as a result of a nitrogen⋯silicon dative bond on each silicon atom. In other words, each nitrogen atom of the azobenzene intramolecularly coordinates to a silicon atom, and its coordination reflects structural perturbation around the silicon atoms. Fluorine atoms increase the electrophilicity of the silicon atoms to a degree high enough to form the N⋯Si dative bonds, overcoming the steric repulsion between the silyl and azo groups. The N⋯Si interatomic distance in (E)-4e (2.371(4) Å) is shorter than that in (E)-4d (2.585(3) Å), reflecting the high electrophilicity of the silicon atom because of the introduction of more fluorine atoms. Such a tendency is also observed in (E)-2-silylazobenzenes (E)-3.13 The N⋯Si interatomic distance in (E)-4e is slightly longer than that in (E)-3a (R1 = R2 = F; N⋯Si, 2.3030(14) Å),13d suggesting that the intramolecular coordination in (E)-2,2′-disilylazobenzenes is weaker than that in the corresponding (E)-2-monosilylazobenzenes and that the coordination is affected by other additional factors. Namely, in (E)-4e, attractive N1*⋯Si1 coordination causes not only a shortening of the N1*⋯Si1 interatomic distance, but also narrowing and widening of C1–N1–N1* and N1–N1*–C1*, respectively, as in (E)-3a, in which the corresponding C–N–N and N–N–C angles are 111.7(1) and 119.9(1)°, respectively.13 Because another coordination, N1⋯Si1* coordination, causes opposite structural changes of the bond angles, the two conflicting N⋯Si interactions could not be too strong. Such a conflict makes the N⋯Si interatomic distance in (E)-4e slightly longer than that in (E)-3. The N⋯Si interactions in (E)-4a,d,e do not show significant changes in NN bond distances (1.255(4)–1.274(4) Å).
N bond against the C1–N1 or C1*–N1* bond, as shown by the C2–C1–N1–N1* torsion angle, which is close to or equal to 0°. Nitrogen atoms occupy the posterior positions of a Si1–F1 bond with the N1*⋯Si1 interatomic distance (2.371(4)–2.585(3) Å) within the sum (3.65 Å) of the van der Waals radii of both elements.16 The F1–Si1–Cn and F1–Si1–Fn′ bond angles are around 100° and narrower than the bond angle for a tetrahedral structure. These structural parameters show a capped tetrahedral coordination sphere at the silicon atoms as a result of a nitrogen⋯silicon dative bond on each silicon atom. In other words, each nitrogen atom of the azobenzene intramolecularly coordinates to a silicon atom, and its coordination reflects structural perturbation around the silicon atoms. Fluorine atoms increase the electrophilicity of the silicon atoms to a degree high enough to form the N⋯Si dative bonds, overcoming the steric repulsion between the silyl and azo groups. The N⋯Si interatomic distance in (E)-4e (2.371(4) Å) is shorter than that in (E)-4d (2.585(3) Å), reflecting the high electrophilicity of the silicon atom because of the introduction of more fluorine atoms. Such a tendency is also observed in (E)-2-silylazobenzenes (E)-3.13 The N⋯Si interatomic distance in (E)-4e is slightly longer than that in (E)-3a (R1 = R2 = F; N⋯Si, 2.3030(14) Å),13d suggesting that the intramolecular coordination in (E)-2,2′-disilylazobenzenes is weaker than that in the corresponding (E)-2-monosilylazobenzenes and that the coordination is affected by other additional factors. Namely, in (E)-4e, attractive N1*⋯Si1 coordination causes not only a shortening of the N1*⋯Si1 interatomic distance, but also narrowing and widening of C1–N1–N1* and N1–N1*–C1*, respectively, as in (E)-3a, in which the corresponding C–N–N and N–N–C angles are 111.7(1) and 119.9(1)°, respectively.13 Because another coordination, N1⋯Si1* coordination, causes opposite structural changes of the bond angles, the two conflicting N⋯Si interactions could not be too strong. Such a conflict makes the N⋯Si interatomic distance in (E)-4e slightly longer than that in (E)-3. The N⋯Si interactions in (E)-4a,d,e do not show significant changes in NN bond distances (1.255(4)–1.274(4) Å).
Structure in solution
A weak N⋯Si coordination may dissociate in solution even if coordination exists in the crystalline state.15 The 1H, 13C, 19F and 29Si NMR spectra of (E)-4a–e were recorded for determination of their structures in solution. The magnitude of the contribution of the N⋯Si coordination in solution can be estimated by the 29Si NMR chemical shifts, which reflect the coordination state of the silicon atoms. The 29Si NMR chemical shifts of (E)-4a–e in CDCl3 are summarized together with those of the corresponding silylbenzenes and differences (Δδ) between their chemical shifts in Table 2. Compounds (E)-4a–c show chemical shifts that are almost the same as those of the corresponding silylbenzenes, with the difference being less than 1 ppm, indicating the tetracoordination states of the silicon atoms. In contrast, compounds (E)-4d,e resonated upfield by 14–15 ppm. Such chemical shifts in (E)-4d,e indicate the pentacoordination states of the silicon atoms also in solution. It is ascribed to the N⋯Si coordination, which is also found in the crystalline state.Photophysical properties
The photophysical properties of the (E)-4,4′-dimethyl-2,2′-disilylazobenzenes (E)-4a–e were studied using UV-vis absorption spectroscopy. The UV-vis absorption spectra of (E)-4a–e in hexane are shown in Fig. 2. The absorption maxima of (E)-4a–e and (E)-4,4′-dimethylazobenzene ((E)-5) are summarized in Table 3. Compounds (E)-4a–c exhibited two absorption bands, a strong band in the short-wavelength region and a weak band in the long-wavelength region. The bands in the short- and long-wavelength regions are assigned to the allowed π,π* transition and the forbidden n,π* transition of the azobenzene moiety, respectively. Both of the bands are bathochromically shifted from the corresponding bands of non-silylated azobenzene (E)-5. The bathochromic shifts are due to the inductive effect of the electron-donating silyl groups.13,15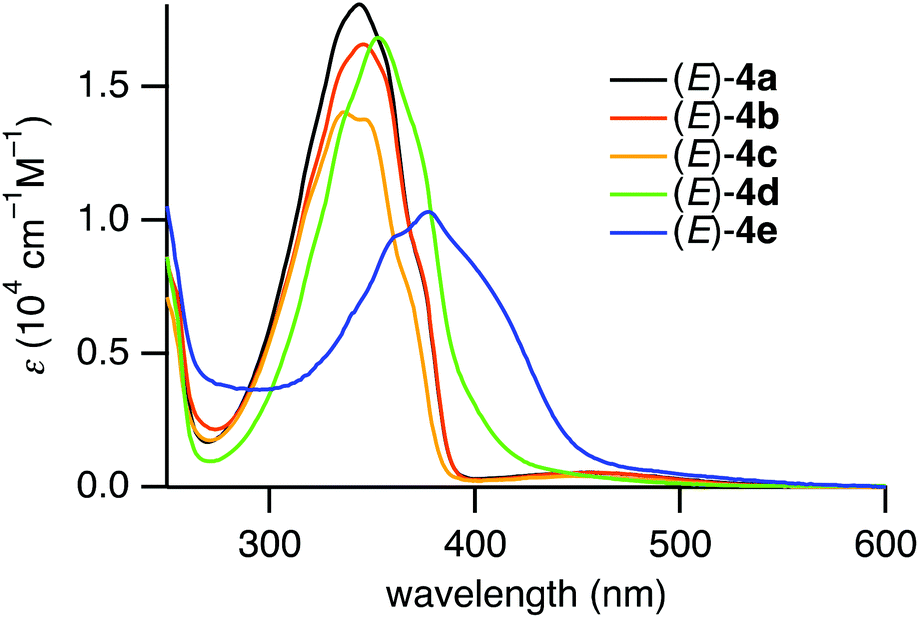 | ||
| Fig. 2 UV-vis absorption spectra of (E)-4a–e in hexane. | ||
In contrast, (E)-4d,e in the coordinating state exhibited a strong band with shoulder peaks and no other independent band. The band ascribed to the π,π* transition is much more bathochromically shifted than the bands of (E)-4a–c. The absorption maxima for the n,π* transition were not observed because their hypsochromic shifts would have caused their overlap with the strong π,π* absorption.15
Irradiation of a hexane solution of (E)-4a–d with UV light from a mercury lamp (λ = 365 nm) resulted in a decrease in the absorbance of the π,π* transition together with an increase of the absorbance of the n,π* transition (see ESI†). Subsequent irradiation of the reaction mixture with visible light (λ = 436 nm) recovered the strong π,π* absorbance of (E)-4a–d. These spectral changes correspond to reversible E,Z-photoisomerization of azobenzene in (E)-4a–d. The E/Z ratio at the photostationary state of a CDCl3 solution of (E)-4a–d under UV irradiation (λ = 365 nm) was determined to be 11/89, 2/98, 40/60 and 7/93, respectively, using 1H NMR spectroscopy (see ESI†).
Similar to (E)-4a–d, exposure of a hexane solution of (E)-4e to UV light (λ = 365 nm) resulted in a decrease in the absorbance of the π,π* transition at 384 nm, which was recovered by irradiating the solution with visible light (λ = 436 nm). The UV-vis spectral changes with isosbestic points during the photoirradiation are shown in Fig. 3. These spectral changes correspond to reversible E,Z-photoisomerization of azobenzene in (E)-4e. The E/Z ratio at the photostationary state under UV irradiation (λ = 365 nm) of a hexane solution was determined to be 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21 using 19F NMR spectroscopy (see ESI†).
:21 using 19F NMR spectroscopy (see ESI†).
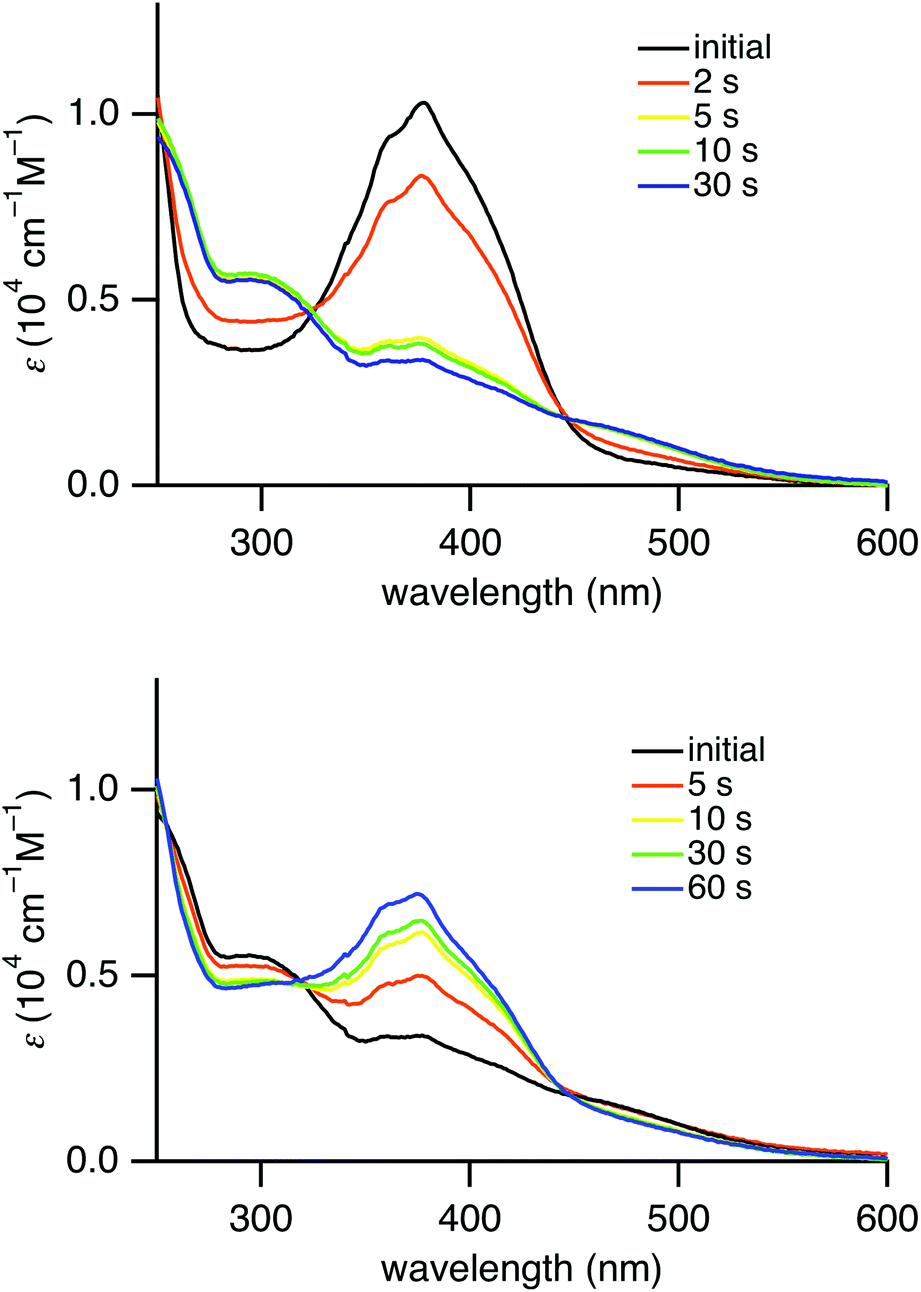 | ||
| Fig. 3 Change of UV-vis absorption spectra of (E)-4e in hexane upon photoirradiation with a mercury lamp (λ = 365 nm) (above) and upon photoirradiation (λ = 436 nm) (below). | ||
Interestingly, (E)-4e in hexane exhibited yellow-green fluorescence at 568 nm at room temperature (Fig. 4). The quantum yield of (E)-4e was determined to be 0.060 in chloroform by an integrating sphere method. Its quantum yield in hexane was similarly determined to be 0.024. These values are significantly larger than the 2.53 × 10−5 for the unsubstituted azobenzene (PhNNPh).19 Although the fluorescence quantum yield is much smaller than the 0.73 for (E)-2,2′-diborylazobenzene (E)-2 (R′ = Bu),12 the fluorescence emission of (E)-4e is worth mentioning because most azobenzene derivatives, including (E)-2-silylazobenzenes, (E)-3, do not fluoresce at all. Considering the photophysical properties, (E)-2,2′-disilylazobenzene (E)-4e was found to be an azobenzene derivative that gives rise to photoisomerization in the excited state and fluorescence from the azo moiety at room temperature.
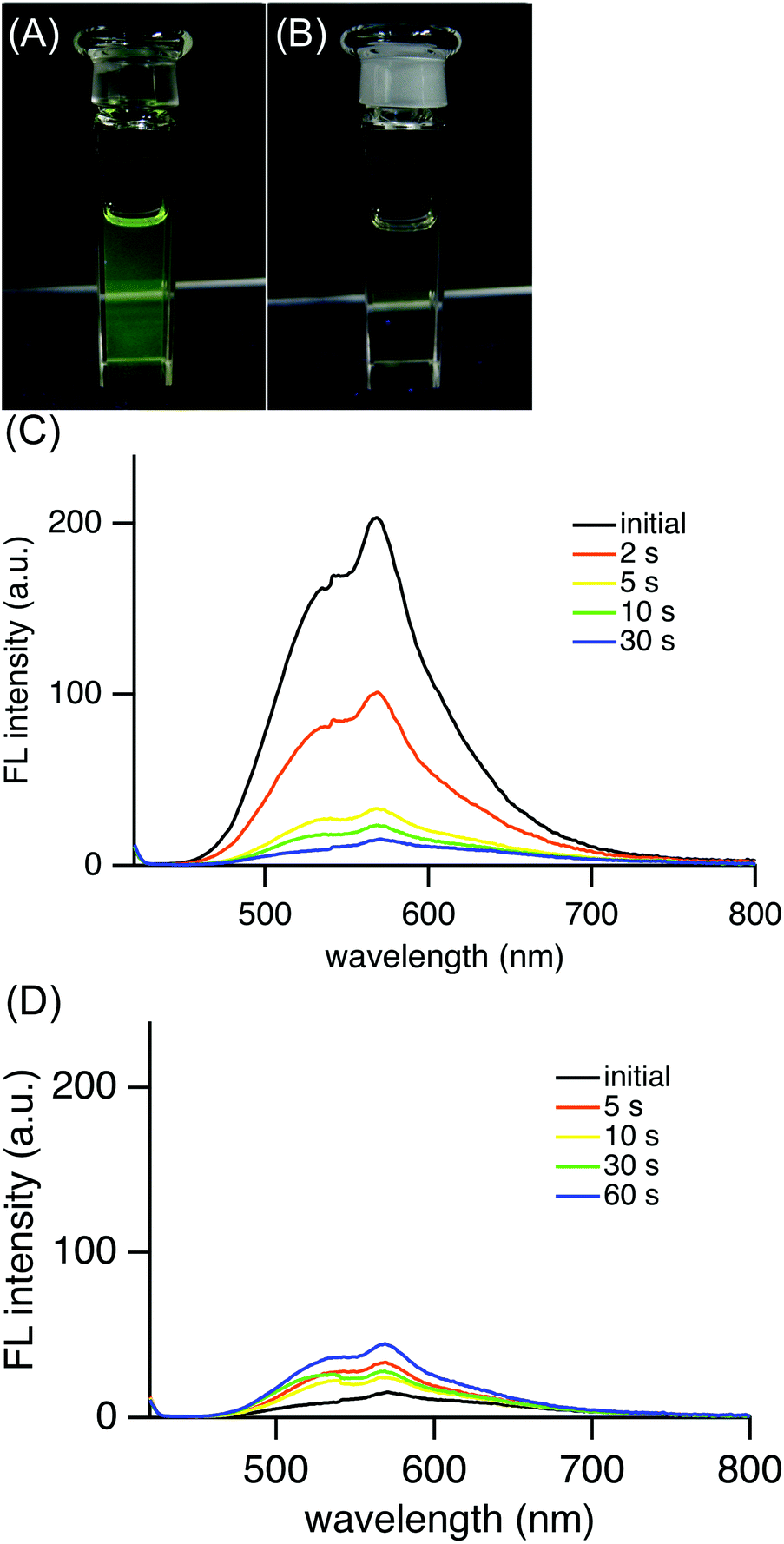 | ||
| Fig. 4 Photograph of (E)-4e in hexane under photoirradiation in the dark (mercury lamp, 400 W, λ = 365 nm) (A) and after irradiation for a minute (B), change of fluorescence spectra (λex = 380 nm) of (E)-4e in hexane upon photoirradiation (λ = 365 nm) (C) and change of fluorescence spectra upon photoirradiation (λ = 436 nm) (D). | ||
Upon photoirradiation, as the fraction of the (E)-isomer of 4e in a mixture of (E)- and (Z)-isomers of 4e decreased, the fluorescence intensity weakened (Fig. 4(C)). Finally, at the photostationary state, the mixture fluoresced very weakly, to the extent of almost non-fluorescence. The fluorescent yellow-green colour of the solution changed to non-fluorescent pale yellow (Fig. 4(A) and (B)). This result can be explained by supposing that (Z)-4e formed from fluorescent (E)-4e does not fluoresce at all. The photoisomerization decreased the fraction of (E)-4e so that the fluorescence intensity also decreased in proportion to the fraction of (E)-4e. After a mixture of (E)- and (Z)-4e had reached the photostationary state with a light of λ = 365 nm, the solution was irradiated with a light of λ = 436 nm and the fluorescence intensity increased in a reflection of the (E)/(Z) ratio at another photostationary state upon irradiation (Fig. 4(D)).
Structure of (Z)-2,2′-disilylazobenzenes
The photophysical properties of the (Z)-isomer are considered to be related to its structure. Taking into account the high E,Z-photoisomerization efficiency, which facilitates both the characterization and isolation of the (Z)-isomer, the structure of (Z)-4d was studied using 29Si NMR spectroscopy and single-crystal X-ray crystallography. The 29Si NMR spectrum of (Z)-4d in CDCl3 showed a doublet at a chemical shift of 22.2 ppm with a coupling constant of 274 Hz. The chemical shift, which is close to that (δ 19.8 ppm) of fluorodimethylsilylbenzene, suggests the absence of the N⋯Si coordination of (Z)-4d, similar to (Z)-3. Recrystallization of (Z)-4d in the dark gave single crystals, which were used for the single-crystal X-ray crystallography. The crystal structure of (Z)-4d depicted in Fig. 5 clearly shows the (Z)-conformation of azobenzenes and the lack of N⋯Si coordination. The N1–N2 bond length (1.256(2) Å) is reasonable for an azobenzene. Thus, it is reasonable that fluorescence was suppressed severely in the mixture containing a certain amount of the (Z)-isomer after the photoisomerization, if the presence of the N⋯Si interaction is a key factor for the fluorescence emission.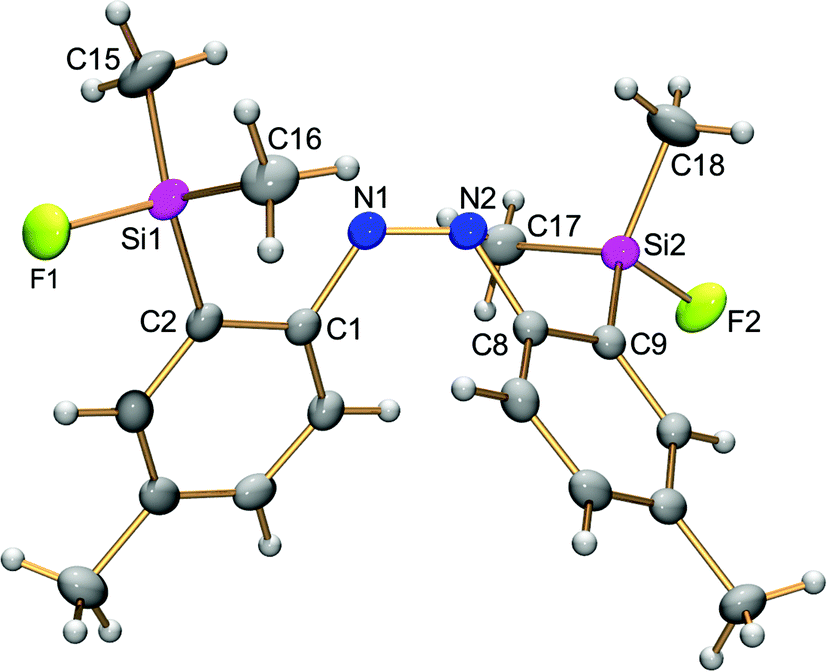 | ||
| Fig. 5 Thermal ellipsoid plot (50% probability) of (Z)-2,2′-disilylazobenzene (Z)-4d. Selected bond lengths (Å), angles (°) and torsion angles (°): N1–N2, 1.256(2); Si1–F1, 1.611(1); Si2–F2, 1.610(1); C1–N1–N2, 123.5(1); N1–N2–C8, 122.5(1); F1–Si1–C2, 103.57(6); F1–Si1–C15, 106.21(8); F1–Si1–C16, 106.50(8); C1–N1–N2–C8, −8.7(2). | ||
Theoretical calculations
Theoretical calculations were performed to shed light on the effects of the N⋯Si coordination on the optical properties of (E)-4e. The structures of (E)-4,4′-dimethyl-2,2′-disilylazobenzene (E)-4e, non-silylated (E)-4,4′-dimethylazobenzene ((E)-5) and (E)-4,4′-dimethyl-2-(trifluorosilyl)azobenzene ((E)-6) were optimized using density functional theory (DFT) calculations at the B3PW91/6-31G(d,p) level. The optimized structures of (E)-4e and (E)-5, whose structures were confirmed by single-crystal X-ray crystallography,20 reproduced the corresponding crystal structures very well. For example, the N–N bond lengths and N⋯Si interatomic distances of the optimized structures ((E)-4e, N–N, 1.255 Å, N⋯Si, 2.369 Å; (E)-5, N–N, 1.259 Å) matched well with the corresponding values in the crystal structures ((E)-4e, N–N, 1.255(6) Å, N⋯Si, 2.371(4) Å; (E)-5, N–N, 1.263(2) Å). Energy diagrams of the Kohn–Sham orbitals of (E)-4e, (E)-5 and (E)-6 were obtained at the B3PW91/6-311++G(2d,p) level (Fig. 6). The HOMO and LUMO of (E)-4e, (E)-5 and (E)-6 are composed of π and π* orbitals, respectively, spreading over the azobenzene moiety. The HOMO−1 of (E)-5 and (E)-6 is composed of the n orbital. Thus, the HOMO character of all three compounds is different from that of non-substituted (E)-azobenzene ((E)-Ph–NN–Ph), in which the HOMO is mainly composed of the n orbital. To discuss substituent effects on the molecular orbitals and their energy levels, both methyl and trifluorosilyl groups should be considered. In (E)-5, the energy gap between the π orbital, HOMO (energy level E, −6.20 eV), and the n orbital, HOMO−1 (E, −6.36 eV), is very small (ΔE, 0.16 eV), even though the order has been inverted compared with that in non-substituted azobenzene.
 | ||
| Fig. 6 Energy diagrams of the Kohn–Sham frontier orbitals of (E)-4e (right), (E)-5 (left) and (E)-6 (middle) at the B3PW91/6-31G(d,p)//B3PW91/6-311++G(2d,p) level: white, hydrogen; grey, carbon; blue, nitrogen; yellow-green, fluorine; orange, silicon. | ||
In both (E)-6 and (E)-4e bearing trifluorosilyl groups, the n orbitals are used for the coordination to silicon atoms, and the N⋯Si coordination causes a large stabilization of the orbitals. As a result, the gap between the n orbital and the HOMO, π orbital, becomes large in both (E)-4e and (E)-6 ((E)-4e, ΔE, 1.13 eV; (E)-6, ΔE, 0.81 eV). In addition, it is noteworthy that the p-orbital of the silicon is involved in the LUMO in (E)-4e and (E)-6. This orbital overlap is assigned to σ*–π* conjugation, which is common in some π-conjugated compounds, such as a silole bearing heavier elements.21 The N⋯Si coordination leads to σ*–π* conjugation and a decrease in the LUMO energy level. The LUMO energy level decreases in the order (E)-5, (E)-6 and (E)-4e, reflecting the magnitude of the σ*–π* conjugation. The σ*–π* conjugation in the LUMO accounts for the large energy gaps between the LUMO of (E)-5 and LUMOs of (E)-6 and (E)-4e. Relatively small energy gaps between the HOMO of (E)-5 and HOMOs of (E)-6 and (E)-4e are reasonable because the conjugation is not involved in the HOMO while it is more dominant in the LUMO.
Vertical excitation energies were obtained using time-dependent DFT (TD-DFT) calculations at the B3PW91/6-311++G(2d,p) level based on the optimized structures. Diagrams of the excitation energies of (E)-4e, (E)-5 and (E)-6 are shown in Fig. 7. The transition from the ground state, S0, to the lowest singlet excited state, S1, for (E)-5, which has no silyl group, corresponds to the transition from HOMO−1 to LUMO, and it is assignable to the forbidden n,π* transition (excitation energy E, 2.60 eV; oscillator strength f, 0.0000) (see ESI†). The character of the S0 → S1 transition is the same as that of non-substituted (E)-azobenzene, in which the S0 → S1 transition corresponds to the n,π* transition. The transition between S0 and the second singlet excited state, S2, for (E)-5 corresponds to the transition from HOMO to LUMO, and it is assignable to the π,π* transition (E, 3.54 eV; f, 0.9510). In contrast, the S0 → S1 transition for (E)-4e corresponds to the HOMO → LUMO transition, which is assignable to the allowed π,π* transition (E, 3.02 eV; f, 0.6421), and the S0 → S2 transition corresponds to the HOMO−3 → LUMO transition, which is assignable to the forbidden n,π* transition (E, 3.18 eV; f, 0.0000). In (E)-4e, the singlet excitation energy of the π,π* transition has decreased, reflecting the large stabilization of the π* orbital because of the σ*–π* conjugation. The singlet excitation energy of the n,π* transition has increased, reflecting the stabilization of the n orbital. As a result, the order of the n,π* and π,π* transitions for (E)-4e is inverted from that of (E)-5. These results of the calculations indicate that the coordination of the azo moiety to the silicon atoms greatly contributes to the perturbation of both the electronic structures and excitation energies.
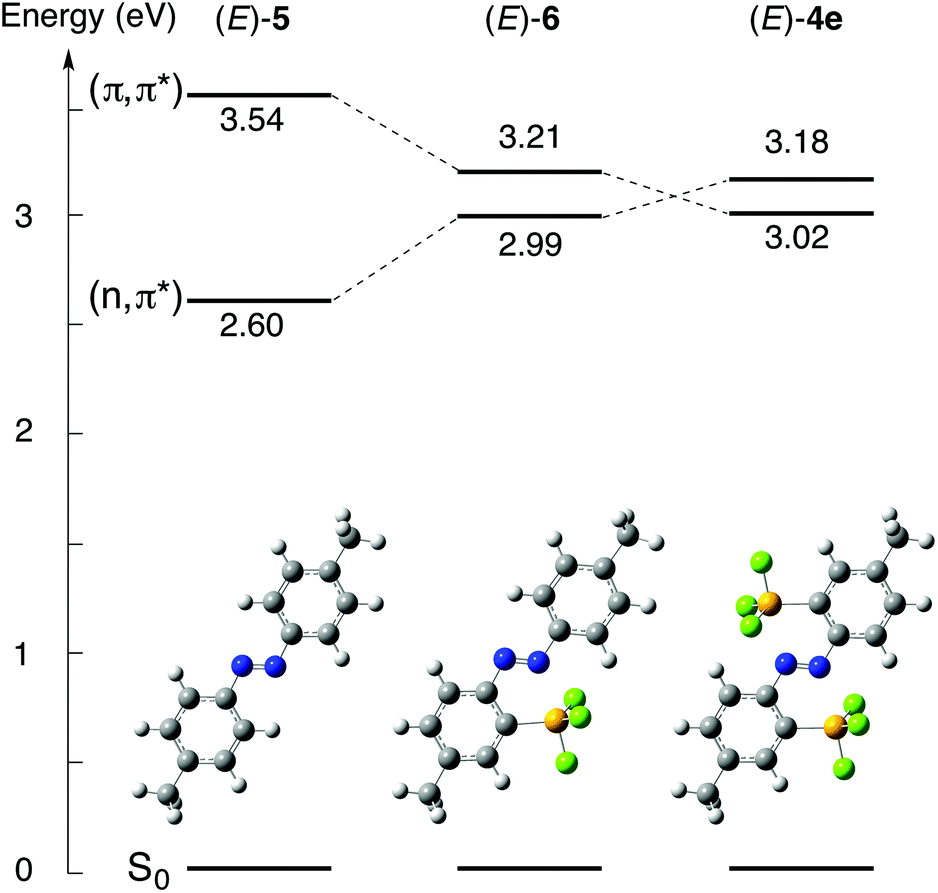 | ||
| Fig. 7 Energy diagrams of the singlet excitation energies of (E)-4e (right), (E)-5 (left) and (E)-6 (middle) at the B3PW91/6-31G(d,p)//B3PW91/6-311++G(2d,p) level together with their optimized structures. | ||
The singlet excitation energy of the π,π* transition (E, 3.21 eV; f, 0.7416) for (E)-4,4′-dimethyl-2-silylazobenzene (E)-6 has decreased significantly from that of (E)-5, but it remains above the increased energy level of the n,π* transition (E, 2.99 eV; f, 0.0009). The single N⋯Si coordination in (E)-6 is found to be too weak to invert the order of the energy levels from that in (E)-5. Therefore, the double coordination to the two silicon atoms in (E)-4e is crucial for the inversion of the order of n,π* and π,π* transitions. The allowed π,π* transition energies decrease in the order (E)-5, (E)-6 and (E)-4e. The π,π* transition energy of (E)-4e being lower than that of (E)-5 is consistent with the bathochromic shift of the corresponding absorption bands in the UV-vis absorption spectrum of (E)-4e.
The results of the theoretical calculations explain why (E)-4e fluoresces. Before giving the explanation, we would like to describe a reason why popular azobenzene derivatives cannot fluoresce. Most (E)-azobenzene derivatives, such as (E)-5, are excited by photoirradiation and slowly relax to the S1 state by internal conversion. They cannot fluoresce because the S1 → S0 transition is the forbidden n,π* transition. Instead, the photoexcitation energy is used for isomerization to the corresponding (Z)-isomer or a non-radiative relaxation such as molecular vibration. In contrast, in (E)-4e, the S1 → S0 transition is not a forbidden n,π* transition, but an allowed π,π* transition owing to the double N⋯Si coordination. Furthermore, the rigid structure because of the interaction prohibits non-radiative relaxation to some extent. Thus, upon photoirradiation, excited (E)-4e can relax from S1 to S0 accompanied by fluorescence emission. This is a reason for the fluorescence emission of (E)-4e.
Although the fluorescence emission of (E)-4e is explained similarly to that of (E)-1 and (E)-2 featuring N⋯B coordination,11,12 we need an answer for the question why (E)-4e photoisomerizes as well as fluoresces. A reason for the photoisomerization properties of (E)-4e is the weaker N⋯Si coordination than that in (E)-2-trifluorosilylazobenzene, as revealed by the X-ray crystallographic analysis. The weak N⋯Si coordination should be cleaved during/after the photoexcitation, and isomerization should occur during the non-radiative decay process. Considering that (E)-2-trifluorosilylazobenzene, which has stronger N⋯Si coordination than (E)-4e, can isomerize upon photoirradiation,13d it is reasonable that (E)-4e can undergo photoisomerization by competing with the fluorescence emission process. The weak N⋯Si coordination of (E)-4e should be a key factor for the bifunctional character, fluorescence emission and photoisomerization, the latter of which cannot be achieved in the fluorescent (E)-2-diborylazobenzenes (E)-1 and (E)-2,2′-diborylazobenzenes (E)-2.
Experimental
General procedure
THF and diethyl ether (Et2O) were distilled from sodium and benzophenone before use. All reactions were carried out in oven-dried glassware under an argon atmosphere. 2,2′-Diiodo-4,4′-dimethylazobenzene was synthesized according to the literature. The 1H NMR (600, 500 and 400 MHz), 13C NMR (150, 126 and 100 MHz) and 29Si NMR (99 MHz) spectra were recorded on JEOL A500 and AL400 or Bruker AVANCE600 spectrometers using tetramethylsilane (TMS) as an internal standard. The 19F NMR (376 MHz) spectra were recorded on a JEOL AL400 spectrometer using Freon as an external standard. All melting points are uncorrected. Elemental analyses were performed by the Microanalytical Laboratory of Department of Chemistry, Faculty of Science, University of Tokyo.Synthesis of (E)-2,2′-disilylazobenzenes
Compounds (E)-2a–c were synthesized from (E)-2,2′-diiodo-4,4′-dimethylazobenzene according to the procedure reported previously in ref. 15.Synthesis of (E)-2,2′-bis(fluorodimethylsilyl)-4,4′-dimethylazobenzene ((E)-4d)
To a THF solution (20 mL) of (E)-4b (144 mg, 0.440 mmol) was added silver(I) fluoride (451.1 mg, 3.55 mmol) at ambient temperature and the reaction mixture was stirred for 20 hours. After the residual salt was filtered off, the filtrate was concentrated. The crude product was purified by preparative HPLC to give yellow crystals of (E)-4d (119 mg, 0.328 mmol, 75%).(E)-4d: yellow crystals; yield 75%; m.p. 179–180 °C; 1H NMR (600 MHz, CDCl3) δ 0.48 (d, 3JF–H = 3.2 Hz, 12H), 2.48 (s, 6H), 7.44 (dd, 3J = 8.4 Hz, 4J = 1.4 Hz, 2H), 7.80 (d, 4J = 1.4 Hz, 2H), 7.89 (d, 3J = 8.4 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 1.36 (d, 2JC–F = 19.2 Hz), 21.62 (s), 125.17 (s), 131.66 (s), 131.73 (d, 2JC–F = 15.7 Hz), 136.71 (d, 3JC–F = 6.8 Hz), 142.11 (s), 152.44 (d, 3JC–F = 2.4 Hz); 19F NMR (376 MHz, CDCl3) δ −149.17 (brs, 1JSi–F = 265.6 Hz); 29Si NMR (99 MHz, CDCl3) δ 4.91 (d, 1JSi–F = 265.6 Hz); UV/vis (hexane) λmax (ε) 353 nm (1.68 × 104); Anal. Calcd for C18H24N2F2Si2: C, 59.63; H, 6.67; N, 7.73. Found: C, 59.84; H, 6.74; N, 7.53%.
Synthesis of (E)-4,4′-dimethyl-2,2′-bis(trifluorosilyl)azobenzene ((E)-4e)
To an ether solution (10 mL) of (E)-4c (54.5 mg, 0.102 mmol) was added boron trifluoride-diethyl ether (50 μL, 0.40 mmol) at ambient temperature and the reaction mixture was stirred for 6 hours. After the reaction mixture was concentrated, dry hexane was added. The insoluble product was filtered off and the filtrate was concentrated. The crude product was purified by recrystallization from hexane to give yellow crystals of (E)-4e (8.1 mg, 0.021 mmol, 21%). Compound (E)-4e was gradually decomposed.(E)-4e: yellow crystals; 1H NMR (600 MHz, C6D6) δ 1.80 (s, 6H), 6.83 (d, 3J = 8.0 Hz, 2H), 7.64 (s, 2H), 8.07 (d, 3J = 8.0 Hz, 2H); 13C{1H} NMR (150 MHz, C6D6) δ 20.80, 129.42, 134.67, 139.97, 145.10 (two aromatic carbons were not observed probably due to the complex coupling with fluorine atoms); 19F NMR (376 MHz, CDCl3) δ −140.72 (s, 1JSi–F = 237.4 Hz); 29Si NMR (99 MHz, CDCl3) δ −87.73 (q, 1JSi–F = 237.4 Hz); UV/vis (hexane) λmax (ε) 378 nm (1.02 × 104).
Typical procedure for photoisomerization of 2,2′-disilylazobenzenes
A CDCl3 solution of (E)-4a (5.8 mM, 0.5 mL) in a Pyrex NMR tube was irradiated with an emission line at 365 nm from a high-pressure mercury lamp (500 W) through a colored-glass filter for two hours at room temperature. In the 1H NMR spectrum, new signals assigned to (Z)-4a were observed. The ratio of (E)-4a and (Z)-4a was determined to be 11:89 by the integral of the NMR signals. Further irradiation at 365 nm did not change the ratio. The reaction mixture was irradiated with an emission line at 436 nm for two hours at room temperature, and then the ratio of (E)-4a and (Z)-4a was changed to be 84:16. Further irradiation at 436 nm did not change the ratio.
(Z)-4a: red solution in CDCl3; E/Z, 11/89 (at the photostationary state, λ = 365 nm), 84/16 (λ = 436 nm); 1H NMR (600 MHz, CDCl3) δ 0.40 (s, 18H), 2.28 (s, 6H), 5.90 (d, 3J = 8.4 Hz, 2H), 6.78 (dd, 3J = 8.4 Hz, 4J = 1.4 Hz, 2H), 7.38 (d, 4J = 1.4 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ −0.04, 21.26, 122.76, 129.01, 129.28, 135.98, 136.35, 156.57; 29Si NMR (99 MHz, CDCl3) δ −3.62 (s).
(Z)-4b: red solution in CDCl3; E/Z, 12/88 (365 nm), 80/20 (436 nm); 1H NMR (500 MHz, CDCl3) δ 0.45 (d, 3J = 3.5 Hz, 12H), 2.21 (s, 6H), 4.60 (sept, 3J = 3.5 Hz, 1JSi–H = 191.5 Hz, 2H), 5.94 (d, 3J = 8.4 Hz, 2H), 6.82 (dd, 3J = 8.4 Hz, 4J = 1.4 Hz, 2H), 7.43 (d, 4J = 1.4 Hz, 2H).
(Z)-4c: red solution in CDCl3; E/Z, 40/60 (365 nm), 80/20 (436 nm); 1H NMR (600 MHz, CDCl3) δ 1.26 (t, 3J = 7.2 Hz, 18H), 2.26 (s, 6H), 4.00 (q, 3J = 7.2 Hz, 12H), 6.19 (d, 3J = 8.4 Hz, 2H), 6.84 (dd, 3J = 8.4 Hz, 4J = 1.4 Hz, 2H), 7.58 (d, 4J = 1.4 Hz, 2H).
(Z)-4d: red solution in CDCl3; E/Z, 7/93 (λ = 365 nm), 83/17 (λ = 436 nm); 1H NMR (500 MHz, CDCl3) δ 0.58 (d, 3JF–H = 7.8 Hz, 12H), 2.32 (s, 6H), 5.98 (d, 3J = 8.4 Hz, 2H), 6.88 (dd, 3J = 8.4 Hz, 4J = 1.4 Hz, 2H), 7.56 (d, 4J = 1.4 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ −0.05 (d, 2JC–F = 15.1 Hz), 21.13 (s), 114.91 (s), 130.57 (s), 133.90 (d, 2JC–F = 12.8 Hz), 135.17 (d, 3JC–F = 3.3 Hz), 137.14 (s), 155.87 (s); 19F{1H} NMR (376 MHz, CDCl3) δ −162.64 (s, 1JSi–F = 273.6 Hz); 29Si NMR (99 MHz, CDCl3) δ 22.19 (d, 1JSi–F = 273.6 Hz).
(Z)-4e: red solution in hexane; E/Z, 21/79 (λ = 365 nm), 40/60 (λ = 436 nm); 1H NMR (500 MHz, CDCl3) δ 2.37 (s, 6H), 6.17 (d, 3J = 8.0 Hz, 2H), 7.10 (d, 3J = 8.0 Hz, 2H), 7.74 (s, 2H); 19F NMR (376 MHz, CDCl3) δ −137.49 (s).
X-ray crystallographic analysis of (E)-4a,d,e and (Z)-4d
The X-ray diffraction data were collected on a CCD diffractometer with a graphite-monochromated Mo-Kα radiation. Diffraction data were processed using CrystalClear (Rigaku) for (E)-4a,d and Bruker SAINT (Bruker) for (E)-4e and (Z)-4d. The intensities were corrected for Lorentz and polarization effects. The structures were solved by direct methods (SHELXS-97) and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed at calculated positions and refined isotropically. Crystallographic data of (E)-4a: C20H30N2Si2, MW = 354.64, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.132(5) Å, b = 13.834(9) Å, c = 16.460(11) Å, α = 91.96(2)°, β = 91.53(3)°, γ = 110.682(17)°, V = 2155(2) Å3, Z = 4, μ(Mo-Kα) = 0.169 mm−1, Dc = 1.093 g cm−3, T = 120 K, 7330 measured and 5155 independent reflections, 449 parameters, R1 (I > 2σ(I)) = 0.0769, wR2 (all data) = 0.1651, GOF = 1.112. (E)-4d: C18H24N2F2Si2, MW = 362.57, orthorhombic, Cmca, a = 7.290(5) Å, b = 16.889(12) Å, c = 15.688(12) Å, α = β = γ = 90°, V = 1932(2) Å3, Z = 4, μ(Mo-Kα) = 0.204 mm−1, Dc = 1.247 g cm−3, T = 130 K, 1176 measured and 973 independent reflections, 72 parameters, R1 (I > 2σ(I)) = 0.0473, wR2 (all data) = 0.1286, GOF = 1.117. (Z)-4d: C18H24N2F2Si2, MW = 362.57, triclinic, P, a = 8.9666(5) Å, b = 10.7053(7) Å, c = 11.2276(7) Å, α = 93.383(2)°, β = 106.763(2)°, γ = 99.798(2)°, V = 1010.2(2) Å3, Z = 2, μ(Mo-Kα) = 0.195 mm−1, Dc = 1.192 g cm−3, T = 120 K, 3575 measured and 3359 independent reflections, 224 parameters, R1 (I > 2σ(I)) = 0.0321, wR2 (all data) = 0.0880, GOF = 1.041. (E)-4e: C14H12F6N2Si2, MW = 378.44, monoclinic, P21/c, a = 7.992(5) Å, b = 12.925(8) Å, c = 7.595(5) Å, β = 92.626(7)°, V = 783.7(8) Å3, Z = 2, μ(Mo-Kα) = 0.290 mm−1, Dc = 1.604 g cm−3, T = 120 K, 1706 measured and 1339 independent reflections, 110 parameters, R1 (I > 2σ(I)) = 0.0781, wR2 (all data) = 0.2319, GOF = 1.096. Crystal data are available from the Cambridge Crystallographic Database Centre, deposition no. CCDC 1057523 ((E)-4a), 1057524 ((E)-4d), 1057525 ((E)-4e) and 1057526 ((Z)-4d).
, a = 10.132(5) Å, b = 13.834(9) Å, c = 16.460(11) Å, α = 91.96(2)°, β = 91.53(3)°, γ = 110.682(17)°, V = 2155(2) Å3, Z = 4, μ(Mo-Kα) = 0.169 mm−1, Dc = 1.093 g cm−3, T = 120 K, 7330 measured and 5155 independent reflections, 449 parameters, R1 (I > 2σ(I)) = 0.0769, wR2 (all data) = 0.1651, GOF = 1.112. (E)-4d: C18H24N2F2Si2, MW = 362.57, orthorhombic, Cmca, a = 7.290(5) Å, b = 16.889(12) Å, c = 15.688(12) Å, α = β = γ = 90°, V = 1932(2) Å3, Z = 4, μ(Mo-Kα) = 0.204 mm−1, Dc = 1.247 g cm−3, T = 130 K, 1176 measured and 973 independent reflections, 72 parameters, R1 (I > 2σ(I)) = 0.0473, wR2 (all data) = 0.1286, GOF = 1.117. (Z)-4d: C18H24N2F2Si2, MW = 362.57, triclinic, P, a = 8.9666(5) Å, b = 10.7053(7) Å, c = 11.2276(7) Å, α = 93.383(2)°, β = 106.763(2)°, γ = 99.798(2)°, V = 1010.2(2) Å3, Z = 2, μ(Mo-Kα) = 0.195 mm−1, Dc = 1.192 g cm−3, T = 120 K, 3575 measured and 3359 independent reflections, 224 parameters, R1 (I > 2σ(I)) = 0.0321, wR2 (all data) = 0.0880, GOF = 1.041. (E)-4e: C14H12F6N2Si2, MW = 378.44, monoclinic, P21/c, a = 7.992(5) Å, b = 12.925(8) Å, c = 7.595(5) Å, β = 92.626(7)°, V = 783.7(8) Å3, Z = 2, μ(Mo-Kα) = 0.290 mm−1, Dc = 1.604 g cm−3, T = 120 K, 1706 measured and 1339 independent reflections, 110 parameters, R1 (I > 2σ(I)) = 0.0781, wR2 (all data) = 0.2319, GOF = 1.096. Crystal data are available from the Cambridge Crystallographic Database Centre, deposition no. CCDC 1057523 ((E)-4a), 1057524 ((E)-4d), 1057525 ((E)-4e) and 1057526 ((Z)-4d).
Density functional theory calculations
Geometries of compounds (E)-4e, (E)-5, and (E)-6 were fully optimized with density functional theory at the B3PW91/6-31G(d,p)22 level using the Gaussian 09, Revision B.01 program.23 Cartesian coordinates in the optimized geometry of (E)-4e, (E)-5, and (E)-6 are provided in the ESI.† The Kohn–Sham orbitals and vertical excitation energies were calculated at the B3PW91/6-311G++(2d,p) level.Conclusions
In this paper, we described the synthesis of some (E)-4,4′-dimethyl-2,2′-disilylazobenzenes. Their structures were confirmed both in the crystalline and solution states. The existence of at least one fluorine atom on each silicon atom is crucial to form the double intramolecular N⋯Si coordination. They showed the bathochromic shifts of the π,π* transitions in the absorption spectra from that of non-substituted azobenzene. While most azobenzenes do not fluoresce at all, (E)-4,4′-dimethyl-2,2′-disilylazobenzene (E)-4e, which isomerizes reversibly to the (Z)-isomer, fluoresced a yellow-green colour at room temperature. The photoisomerizability accompanying the fluorescence emission properties is remarkably different from hitherto known azobenzene derivatives. The fluorescence emission of (E)-4e is explained by the radiative relaxation because of the allowed π,π* transition from the lowest singlet excited state as a result of the inverted order of the n,π* and π,π* transitions of the azobenzene moiety because of the double N⋯Si coordination. The N⋯Si coordination is strong enough to cause a perturbation of the electronic structure, but weak enough to be cleaved upon photoexcitation. The moderate strength of the double N⋯Si coordination should be important for realization of both the photoisomerization and fluorescence emission properties of the azobenzene moiety. After the photoisomerization of (E)-4e to the corresponding (Z)-isomer, their mixture hardly fluoresces. Such a drastic change in the fluorescence intensity upon photoisomerization will be useful for regulation of the fluorescence properties, and it is a unique photoisomerizable fluorophore to regulate the fluorescence intensity using a single light source.Acknowledgements
The authors acknowledge financial supports of the Grants-in-Aid for Scientific Research on Innovative Areas “Coordination Programming” (no. 22108508) and for Young Scientists (A) (no. 22685005) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- (a) S. Muramatsu, K. Kinbara, H. Taguchi, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 3764 CrossRef CAS PubMed; (b) G. Pace, V. Ferri, C. Grave, M. Elbing, C. von Hänisch, M. Zharnikov, M. Mayor, M. A. Rampi and P. Samorı, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9937 CrossRef CAS PubMed; (c) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC; (d) E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071 CrossRef CAS PubMed.
- (a) N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875 CrossRef CAS PubMed; (b) A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234 CrossRef CAS PubMed; (c) H. M. Dhammika Bandara, T. R. Friss, M. M. Enriquez, W. Isley, C. Incarvito, H. A. Frank, J. Gascon and S. C. Burdette, J. Org. Chem., 2010, 75, 4817 CrossRef PubMed; (d) H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809 RSC; (e) M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756 CrossRef CAS PubMed.
- H. Rau, in Photochromism: Molecules and Systems, ed. H. Durr and H. Bouas-Lauran, Elsevier, Amsterdam, 1990, p. 165 Search PubMed.
- M. Nepras, S. Lunák, Jr., R. Hrdina and J. Fabian, Chem. Phys. Lett., 1989, 159, 366 CrossRef CAS.
- (a) Y. Wakatsuki, H. Yamazaki, P. A. Grutsch, M. Santhanam and C. Kutal, J. Am. Chem. Soc., 1985, 107, 8153 CrossRef CAS; (b) M. Ghedini, D. Pucci, G. Calogero and F. Barigelletti, Chem. Phys. Lett., 1997, 267, 341 CrossRef CAS; (c) M. Ghedini, D. Pucci, A. Crispini, I. Aiello, F. Barigelletti, A. Gessi and O. Francescangeli, Appl. Organomet. Chem., 1999, 13, 565 CrossRef CAS; (d) I. Aiello, M. Ghedini and M. La Deda, J. Lumin., 2002, 96, 249 CrossRef CAS.
- J. Azuma, A. Shishido, T. Ikeda and N. Tamai, Mol. Cryst. Liq. Cryst., 1998, 34, 83 CrossRef PubMed.
- (a) M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951 CrossRef CAS PubMed; (b) M. R. Han and M. Hara, New J. Chem., 2006, 30, 223 RSC; (c) X. Ran, H. Wang, L. Shi, J. Lou, B. Liu, M. Li and L. Guo, J. Mater. Chem. C, 2014, 2, 9866 RSC.
- F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343 CrossRef CAS PubMed.
- (a) M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175 CrossRef CAS; (b) A. Jacquart, R. M. Williams, A. M. Brouwer and E. Ishow, Chem. – Eur. J., 2012, 18, 3706 CrossRef CAS PubMed.
- S. Tsuchiya, J. Am. Chem. Soc., 1998, 121, 48 CrossRef.
- (a) J. Yoshino, N. Kano and T. Kawashima, Chem. Commun., 2007, 559 RSC; (b) J. Yoshino, N. Kano and T. Kawashima, Chem. Lett., 2008, 37, 960 CrossRef CAS; (c) J. Yoshino, A. Furuta, T. Kambe, H. Itoi, N. Kano, T. Kawashima, Y. Ito and M. Asashima, Chem. – Eur. J., 2010, 16, 5026 CrossRef CAS PubMed; (d) H. Itoi, T. Kambe, N. Kano and T. Kawashima, Inorg. Chim. Acta, 2012, 381, 117 CrossRef CAS PubMed; (e) J. Yoshino, N. Kano and T. Kawashima, Dalton Trans., 2013, 42, 15826 RSC.
- N. Kano, A. Furuta, T. Kambe, J. Yoshino, Y. Shibata, T. Kawashima, N. Mizorogi and S. Nagase, Eur. J. Inorg. Chem., 2012, 1584 CrossRef CAS PubMed.
- (a) N. Kano, F. Komatsu and T. Kawashima, J. Am. Chem. Soc., 2001, 123, 10778 CrossRef CAS; (b) N. Kano, M. Yamamura, F. Komatsu and T. Kawashima, J. Organomet. Chem., 2003, 686, 192 CrossRef CAS; (c) N. Kano, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2004, 126, 6250 CrossRef CAS PubMed; (d) N. Kano, F. Komatsu, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2006, 128, 7097 CrossRef CAS PubMed; (e) M. Yamamura, N. Kano and T. Kawashima, Tetrahedron Lett., 2007, 48, 4033 CrossRef CAS PubMed; (f) M. Yamamura, N. Kano and T. Kawashima, J. Organomet. Chem., 2007, 692, 313 CrossRef CAS PubMed.
- (a) Y. Tokoro, H. Yeo, K. Tanaka and Y. Chujo, Chem. Commun., 2012, 48, 8541 RSC; (b) Q. Xiao, X. Meng, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2014, 53, 3168 CrossRef CAS PubMed; (c) T. Wakaki, M. Kanai and Y. Kuninobu, Org. Lett., 2015, 17, 1758 CrossRef CAS PubMed.
- M. Yamamura, N. Kano, T. Kawashima, T. Matsumoto, J. Harada and K. Ogawa, J. Org. Chem., 2008, 73, 8244 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) C. Brelière, F. Carré, R. J. P. Corriu, M. Poirier and G. Royo, Organometallics, 1986, 5, 388 CrossRef; (b) E. A. Williams and J. D. Cargioli, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, New York, 1979, vol. 9, p. 221 Search PubMed.
- S. Yamamoto, N. Nishimura and S. Hasegawa, Bull. Chem. Soc. Jpn., 1971, 44, 2018 CrossRef CAS.
- T. Fujino, S. Y. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123 CrossRef CAS.
- J. Harada, K. Ogawa and S. Tomoda, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 662 Search PubMed.
- (a) S. Yamaguchi and K. Tamao, Bull. Chem. Soc. Jpn., 1996, 69, 2327 CrossRef CAS; (b) S. Yamaguchi and K. Tamao, J. Chem. Soc., Dalton Trans., 1998, 3693 RSC.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectral charts of (E)-4a–e and (Z)-4a,d,e, UV-vis spectra of (E)- and (Z)-4a–e, fluorescence spectra of (E)- and (Z)-4e, and TD-DFT calculations of (E)-4e, (E)-5, and (E)-6. CCDC 1057523–1057526. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02038g |
| ‡ Present address: Graduate School of Science and Technology, Gunma University, Gunma 376-8515, Japan. E-mail: kawashima.t@gunma-u.ac.jp |
| This journal is © The Royal Society of Chemistry 2015 |