 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent applications of Cp2TiCl in natural product synthesis
Sara P.
Morcillo
,
Delia
Miguel
,
Araceli G.
Campaña
,
Luis
Álvarez de Cienfuegos
,
José
Justicia
and
Juan M.
Cuerva
*
Department of Organic Chemistry, University of Granada, Fuentenueva Campus, Granada 18071, Spain. E-mail: jmcuerva@ugr.es
First published on 27th January 2014
Abstract
This review highlights the recent applications of titanocene(III) complexes in the field of natural product synthesis from the seminal precedents to the development of modern catalytic methods. The power of the titanocene(III)-based approaches is demonstrated by the straightforward syntheses of many natural products from readily available starting materials.
 Jose Justicia, Luis Alvarez de Cienfuegos, Delia Miguel, Juan M. Cuerva, Sara P. Morcillo, Araceli G. Campaña | José Justicia was born in Granada (Spain) in 1975. He obtained his Ph.D. degree at the University of Granada (2004) supervised by Prof. J. M. Cuerva. He performed a postdoctoral stay in the group of Prof. Andreas Gansäuer at the University of Bonn, granted by the Ministerio de Educación y Ciencia (2004–2006). From 2006 to 2011, he had contracts from the “Juan de la Cierva” Program and the “Reincorporacion de Doctores” Program from University of Granada. In 2011, he was promoted to the position of Associate Professor in the Department of Organic Chemistry at that University. Dr Luis Álvarez de Cienfuegos Rodríguez got his Ph.D. degree in Organic Chemistry at the University of Granada in 2003, under the supervision of Prof. Rafael Robles. From 2004 to 2006, he worked as a Fundación Ramon-Areces fellow at MIT in Prof. Alexander M. Klibanov's group. From 2006 to 2007, he worked as a postdoctoral associate at Tufts University in Prof. Krishna Kumar's group. In 2008, he joined, as a researcher, Juan M. Cuerva's group at the University of Granada to work on organometallics and molecular electronics. In 2011, he was promoted to the position of Associate Professor in the Department of Organic Chemistry at that University. His research interests are free radical and transition metal chemistry. Delia Miguel was born in Burgos (Spain) in 1981. She carried out her Ph.D. studies at the University of Burgos (2005–2009) supervised by Prof. Roberto Sanz. During this time she spent a trimester under the supervision of Prof. S. V. Ley at the University of Cambridge (United Kingdom). Her Ph.D. was focused on new strategies for the functionalization of indoles and carbonyl compounds catalyzed by Brønsted acids and gold(I) complexes. Nowadays she is performing a postdoctoral stay in the group of Juan Manuel Cuerva at the University of Granada working on different synthetic applications of multimetallic systems and on the development of organic devices for their use in molecular electronics. Juan M. Cuerva was born in Granada, Spain. He obtained his Ph.D. in 1997 at the Universidad Autónoma de Madrid (Madrid, Spain) under the guidance of Professor Antonio M. Echavarren. In 1998, he moved to the University of Granada and since 2003 he has held the position of Associate Professor of organic chemistry in the Department of Organic Chemistry at that University. He and his research group work on the development of new synthetic methods based on radical chemistry and their applications, especially in the synthesis of natural products. Sara P. Morcillo was born in Granada (Spain) in 1985. She studied Chemistry at Universidad of Granada where she is carrying out her Ph.D. studies on synthesis of natural products under the guidance of Dr José Justicia and Dr Juan M. Cuerva. During this time she has spent a trimester in the lab of Prof. Andreas Gansaüer at the University of Bonn (Germany). Araceli G. Campaña obtained her Ph.D. degree at the University of Granada in 2008, guided by Prof. J. M. Cuerva. Her Ph.D. thesis dealt with the development of new synthetic methods based on multimetallic systems. Subsequently, she completed a postdoctoral stage in the lab of Prof. D. J. Cárdenas at Universidad Autónoma de Madrid (2009). From 2010 to 2012, she worked in the group of Prof. D. A. Leigh at the University of Edinburgh as a postdoctoral fellow studying small-molecule walker systems. Nowadays she is working as a ‘Juan de la Cierva’ postdoctoral fellow in Prof. Cuerva's group. |
Introduction
Natural product synthesis is an exigent test for newly developed methodologies. Within this context, Rajanbabu and Nugent reported a series of seminal papers about the potential role of Cp2TiCl as a new tool in organic synthesis.1 Soon afterwards, Gansäuer's group published a collection of relevant papers where a substoichiometric version of this protocol was developed.2 Those results were especially important in the development of the corresponding asymmetric reactions using chiral titanocene(III) complexes.3 After these inspiring works, titanocene(III) complexes, essentially titanocene(III) chloride (Cp2TiCl), have recently emerged as a powerful tool in organic synthesis. They are soft single-electron-transfer (SET) reagents capable of promoting different kinds of reactions, such as homolytic epoxide4 and oxetane5 openings, Barbier-type reactions,6 Wurtz-type reactions,7 Reformatsky-type reactions,8 reduction reactions,9 and pinacol coupling reactions (Scheme 1).10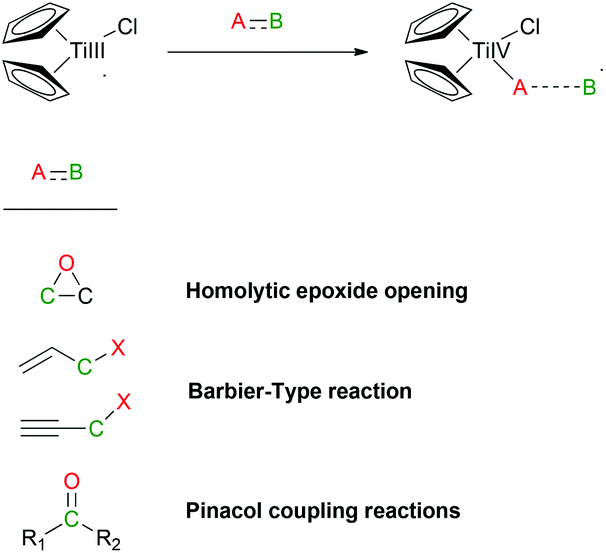 | ||
| Scheme 1 General reactivity of Cp2TiCl. | ||
From a practical point of view, titanocene(III) complexes can be prepared and stored. Nevertheless, they are usually highly oxygen-sensitive compounds. Interestingly, they can be easily prepared in situ by simply stirring the corresponding titanocene(IV) precursor and manganese or zinc dust. Another key characteristic of the titanocene(III) chemistry is that whatever the reaction in which it is involved, a catalytic cycle can be closed. In that case, a titanocene(IV) regenerating agent and an electron source, such as manganese or zinc dust, are required. Although some of them have been described in the literature, only two are commonly used: the simple combination of trimethylsilyl chloride and 2,4,6-collidine for aprotic reaction conditions11 and 2,4,6-collidinium hydrochloride for aqueous conditions (see Scheme 2).2
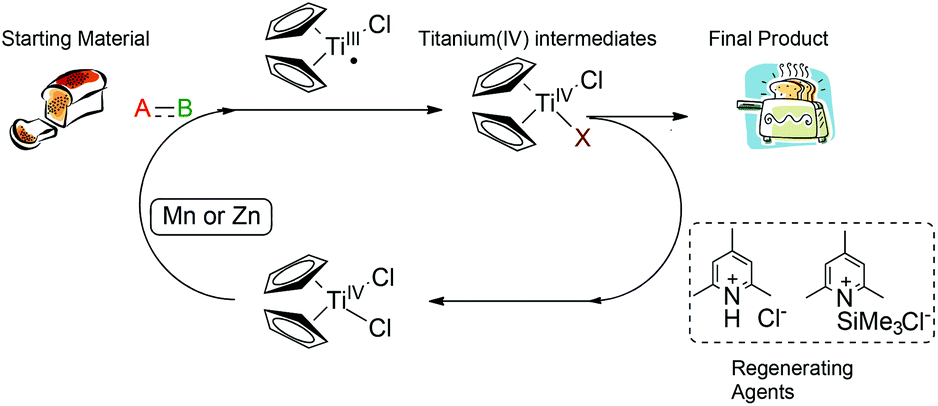 | ||
| Scheme 2 Catalytic cycles in titanocene(III) chemistry. | ||
As far as the starting materials in titanocene(III) chemistry are concerned, they must be activated owing to the modest reduction potential of those complexes, although some exceptions have been reported.12 Therefore, the usual titanocene(III) chemistry is restricted to three big families: epoxides, allylic and propargylic halides and α,β-unsaturated and aromatic aldehydes and ketones (see Scheme 1).
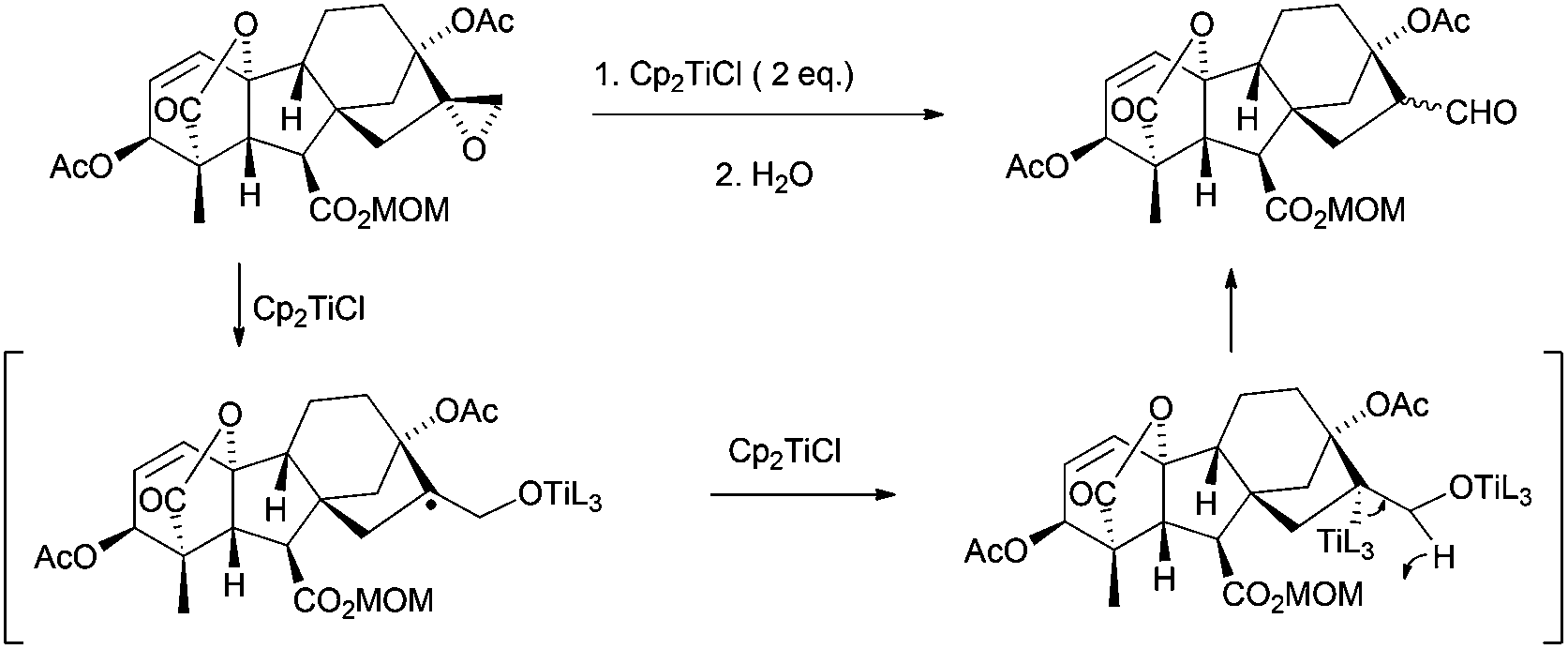
Thus, titanocene(III) complexes can interact with epoxides to yield the corresponding carbon-centred radicals. The fate of these radicals depends upon the reaction conditions. In the presence of an excess of titanocene(III), deoxygenation reaction takes place. In the presence of a good hydrogen-atom donor, such as 1,4-cyclohexadiene, the corresponding alcohols with anti-Markovnikov regioselectivity are obtained. In the presence of an alkene, an alkyne or a carbonyl group a new C–C bond is formed, either inter- or intramolecularly. Another remarkable feature is their reactivity towards substituted carbon-centered radicals. Such radicals are usually transformed into alkenes by a mixed disproportionation process. That is, Cp2TiCl is able to abstract a hydrogen atom of the α-position of a carbon-centered radical to form a C–C double bond and Cp2TiClH. This transformation formally represents quite an unusual oxidation reaction of the organic substrate in radical chemistry (Scheme 3).13
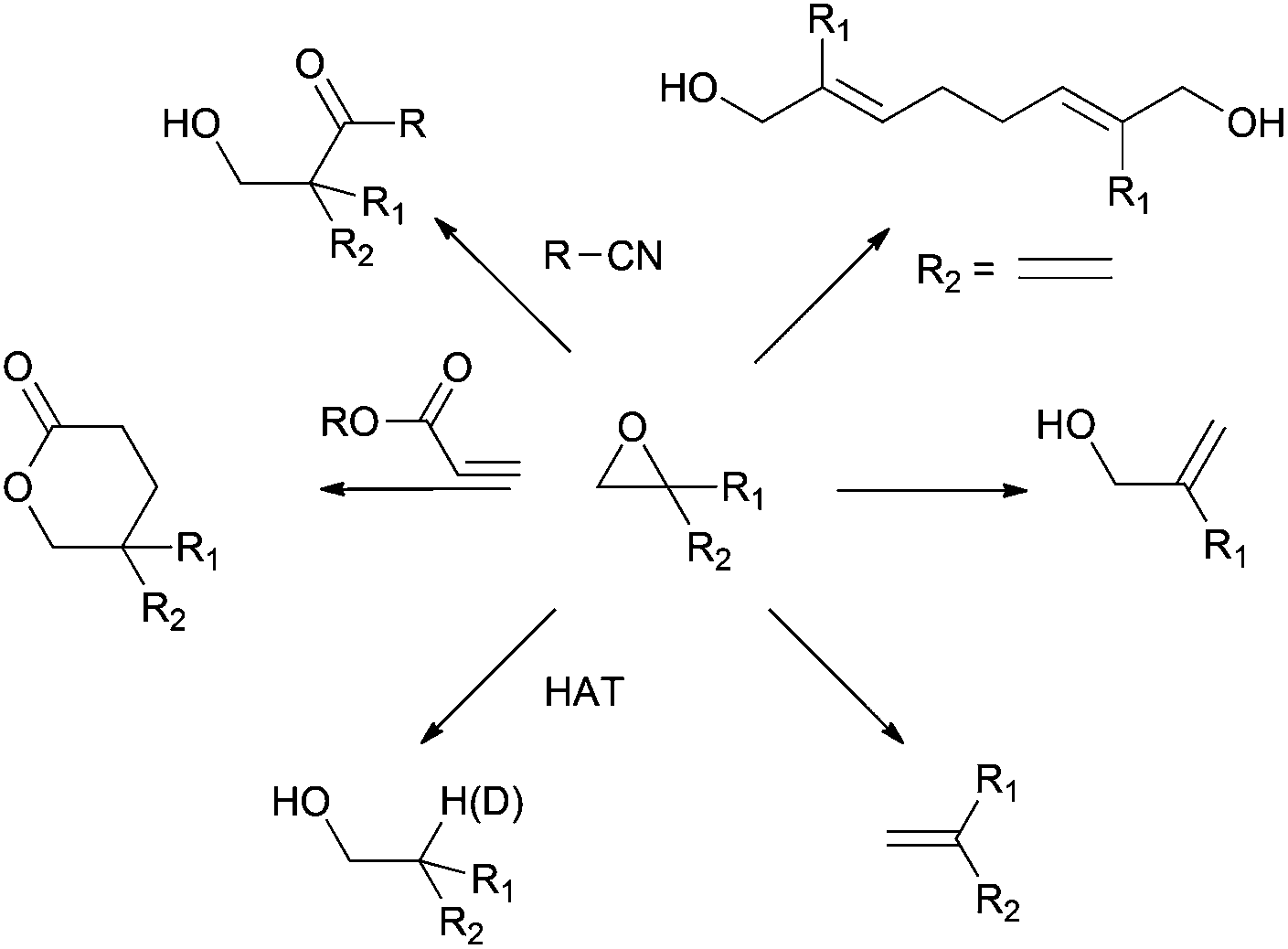 | ||
| Scheme 3 Reactivity of epoxides in titanocene(III) chemistry. | ||
Allylic and propargylic halides have been used in Barbier-type reactions with interesting regio- and chemoselectivities.6 Moreover, excellent D,L-meso stereoselectivities have been obtained in titanocene(III)-mediated pinacol coupling reactions of α,β-unsaturated and aromatic aldehydes and ketones (Scheme 4).10
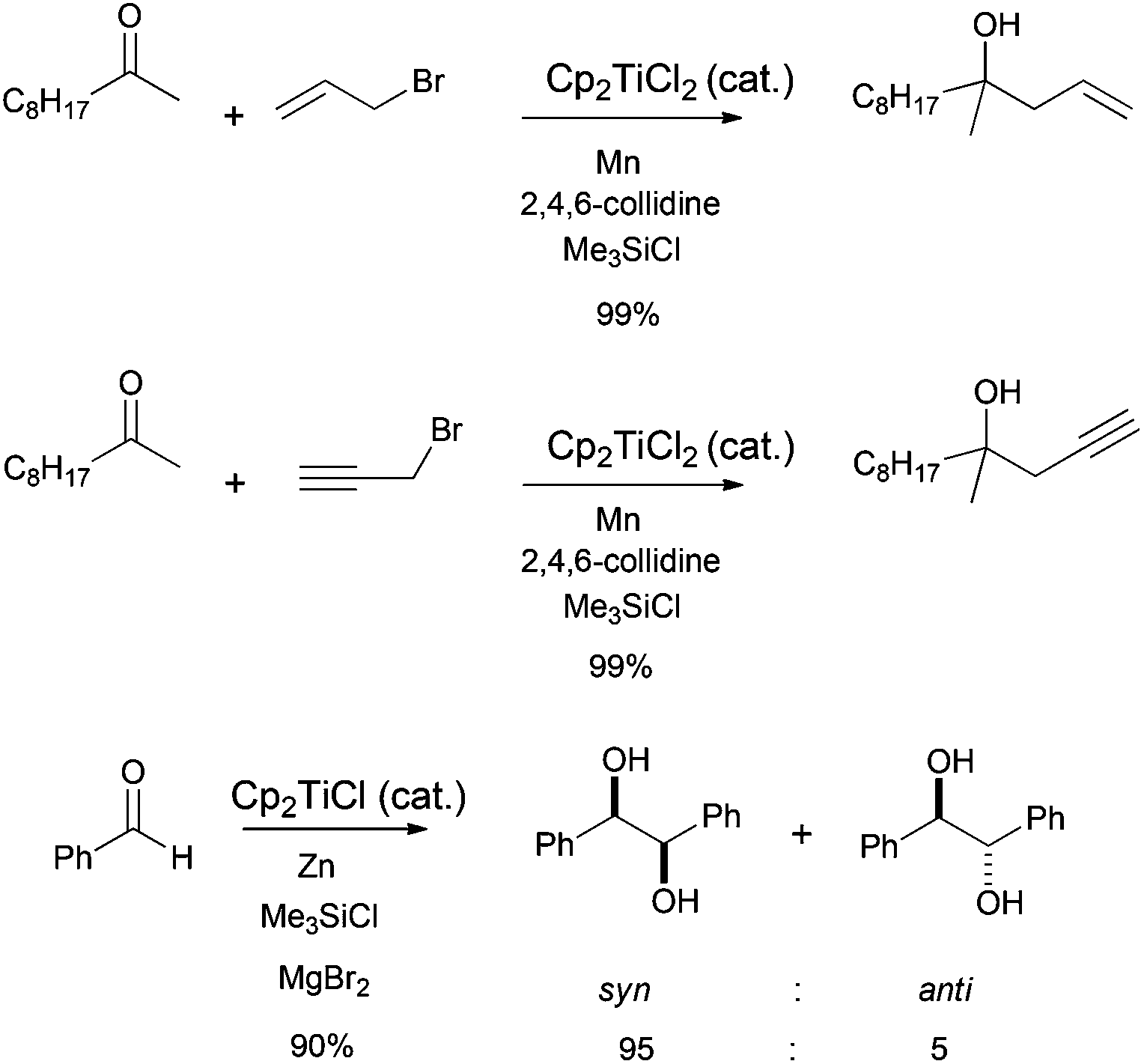 | ||
| Scheme 4 Examples of Cp2TiCl-catalyzed Barbier-type and pinacol coupling reactions. | ||
All these reactions have been applied in functional group interconversions in some natural product synthesis, taking advantage of their remarkable chemoselectivity. More relevant is the fact that titanocene(III)-mediated reactions have been used as the key step in the synthesis of different complex natural products such as sesterstatin 1, barekoxide, or sicannin, showing the potential of this reagent.
In this article we have summarized such contributions in the field of natural products synthesis with special emphasis in recent years.4b,c We have distinguished the role of titanocene(III)-complexes as a simple reagent and their use in building the main framework. Therefore, the article is subdivided into two sections devoted to these different applications.
Useful Cp2TiCl-mediated functional group interconversions in the context of natural product synthesis
As we had commented before, the fate of carbon-centred radicals generated by Cp2TiCl is determined by the reaction conditions employed. Thus, for example, such intermediates can participate in useful reduction or oxidation reactions.Deoxygenations and dehydrogenations
In the treatment of an epoxide with Cp2TiCl, as firstly presented by Rajanbabu et al., the deoxygenation product appears from the reduction of the intermediate β-titanoxy radical by another molecule of Cp2TiCl followed by elimination of titanocene(IV) oxides, thus yielding the corresponding olefinic product (Scheme 5).14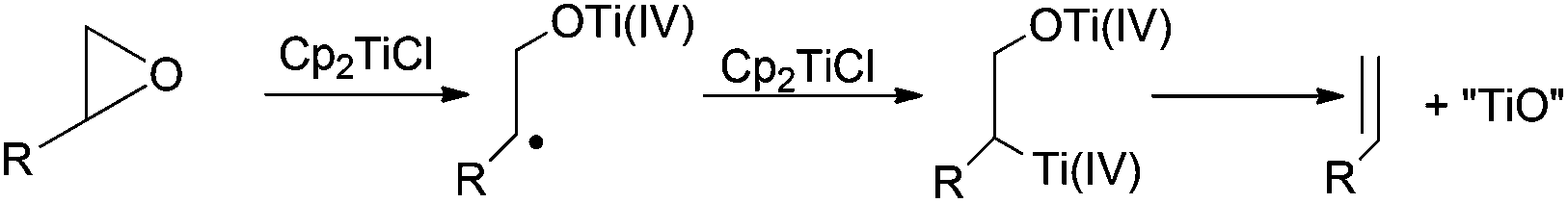 | ||
| Scheme 5 General mechanism for epoxide deoxygenation. | ||
This methodology showed an interesting value and was applied to the synthesis of several cryptophycin derivatives within the context of a structure–activity relationship study for their antitumor evaluation.15 The synthesis of antitumoral alkaloid anhydrovinblastine from the selective deoxygenation of leurosine with Cp2TiCl is also quite remarkable (Scheme 6).16
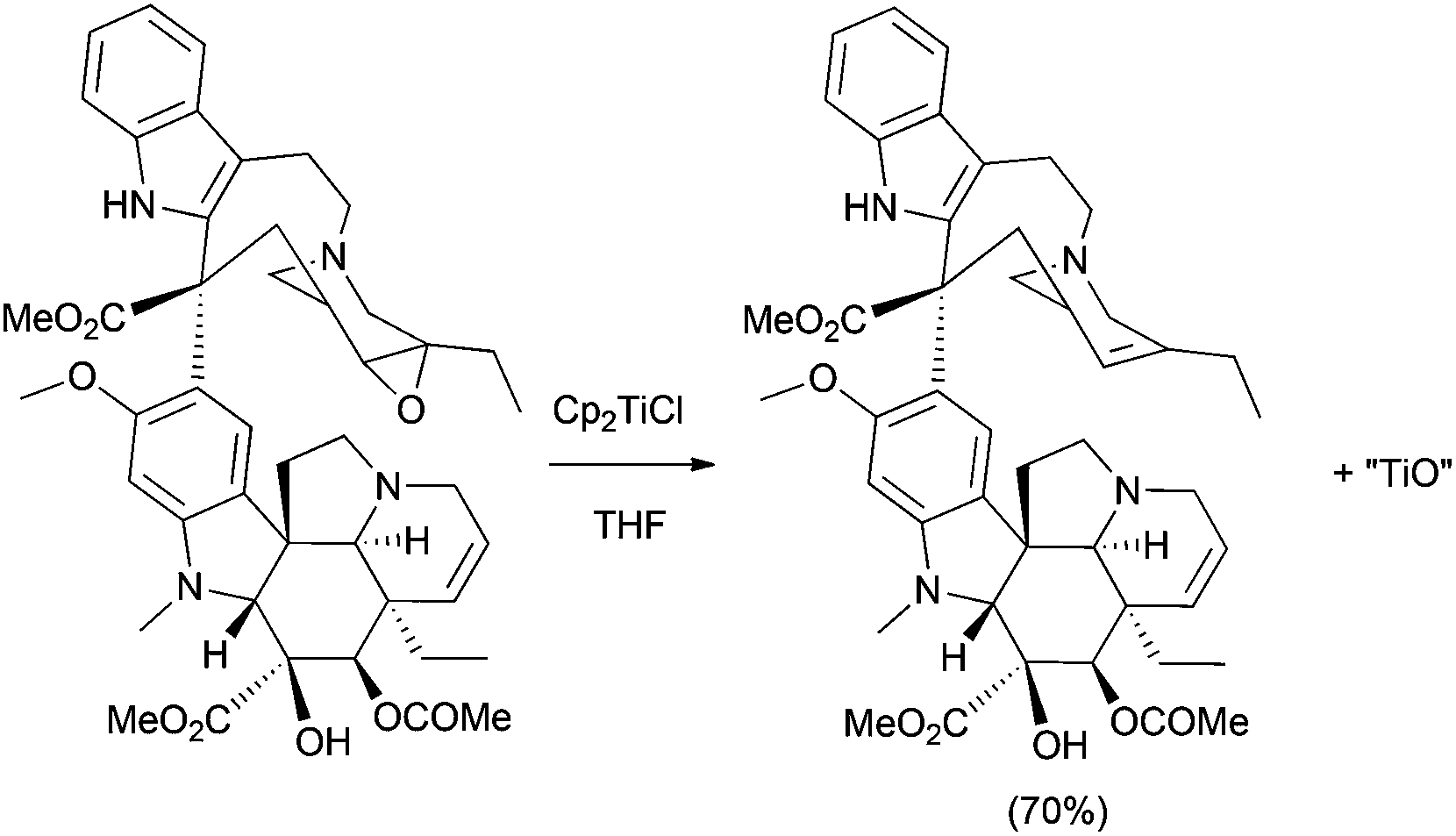 | ||
| Scheme 6 Synthesis of anhydrovinblastine from leurosine. | ||
This useful transformation has also been valuable in carbohydrate chemistry for glycal synthesis from the corresponding glycosyl halides.17 Furthermore, the amount of Cp2TiCl required for glycal formation was considerably lowered to only 30 mol% by combination with a regenerating agent such as Me3SiCl (Scheme 7).18
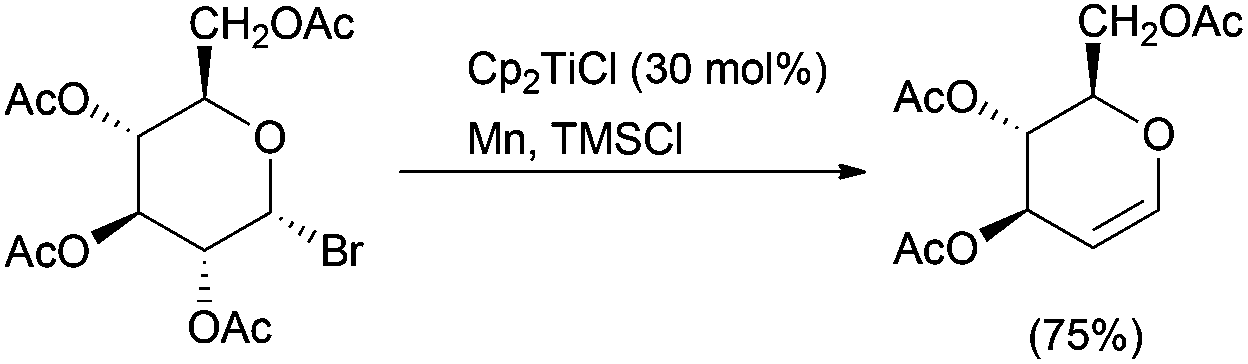 | ||
| Scheme 7 Glycal synthesis. | ||
It was observed by Yavav and co-workers that treatment of 2,3-epoxy alcohols with Cp2TiCl led to the corresponding allylic alcohols by a highly regioselective deoxygenation reaction.19 Following this protocol, the stereoselective synthesis of the C-12 to C-18 segment of antibiotic rhizoxin 1 (Scheme 8) was performed from chiral carbinol 2, prepared by a titanocene-mediated ring opening of epoxy alcohol 3.20
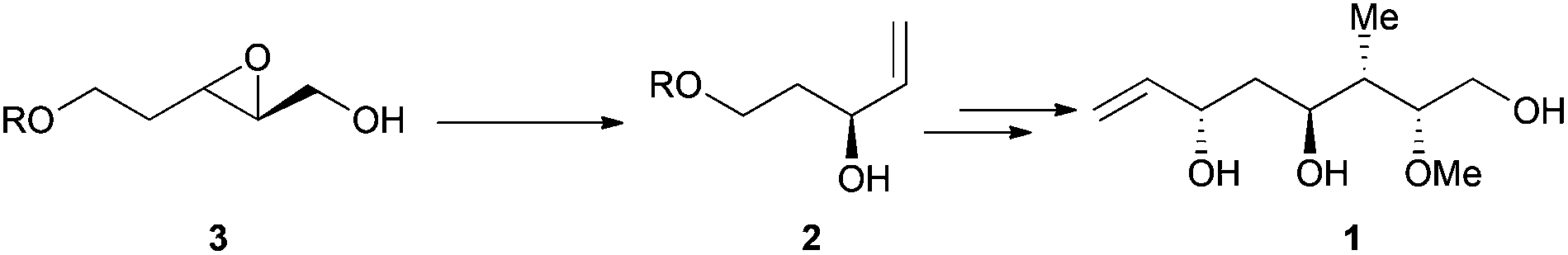 | ||
| Scheme 8 Synthesis C-12 to C-18 fragment 1 of antibiotic rhizoxin. | ||
The same protocol has also been recently applied to prepare a lipid olefin (Scheme 9), which was subsequently used in the synthesis of C-glycoside analogues of α-galactosylceramide.21
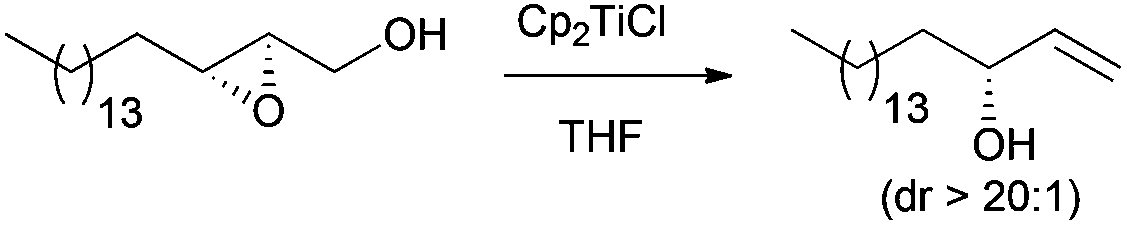 | ||
| Scheme 9 Regioselective deoxygenation of 2,3-epoxy alcohols. | ||
Moreover, Yavav extended the deoxygenation protocol to the treatment of epoxy allylic alcohols with Cp2TiCl, leading to the highly regioselective synthesis of chiral butadienyl alcohols, which are valuable synthons in natural product synthesis (Scheme 10).22
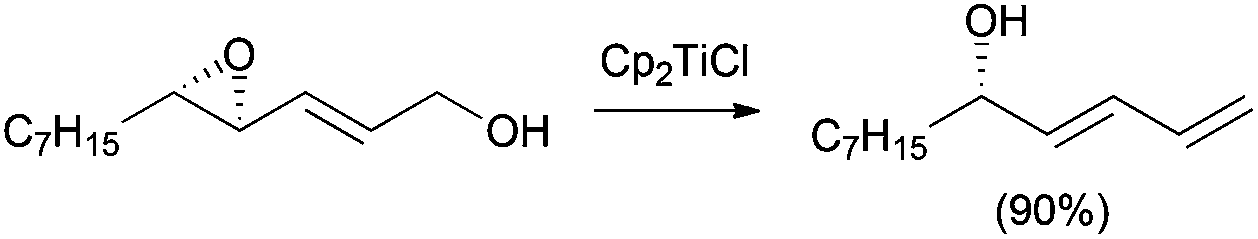 | ||
| Scheme 10 Synthesis of butadienyl alcohols. | ||
In the presence of an excess of Cp2TiCl, primary and secondary carbon-centred radicals undergo the previously described deoxygenation reaction to yield an alkene. Nevertheless, the formation of allylic alcohols is commonly observed in reactions involving tertiary radicals. In this case, an alternative mixed disproportionation process has been suggested to be involved.13 Some representative examples are shown in Scheme 11.
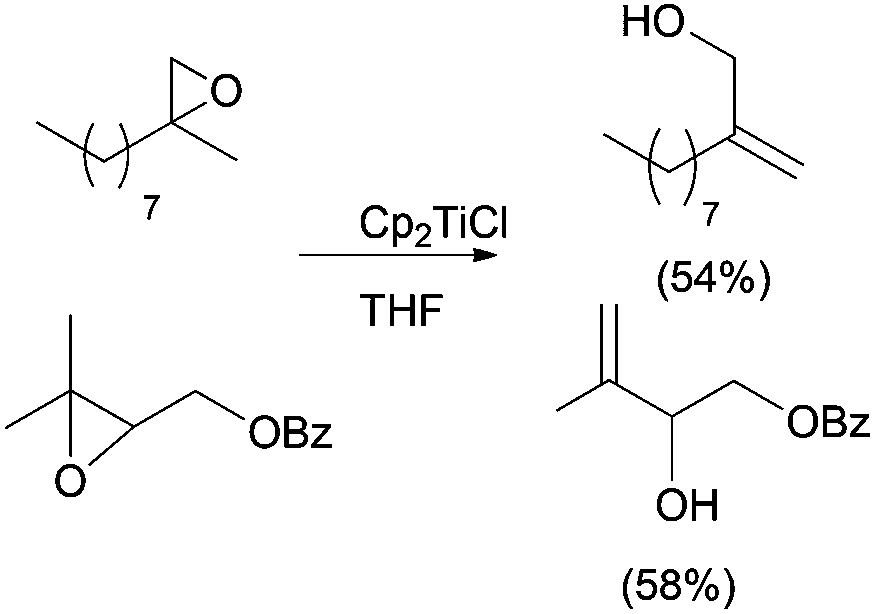 | ||
| Scheme 11 Allylic alcohols obtained from tertiary radicals. | ||
In this sense, ring opening reactions of trisubstituted epoxides on carvone derivatives led to exo-methylene allylic alcohols as the major products (Scheme 12).23 On the other hand, the treatment of α-pinene oxide derivatives with 2 equiv. of Cp2TiCl could selectively afford a series of valuable compounds with an endocyclic double bond in their structure (Scheme 13).24 Moreover, the radical formed after the homolytic cleavage of oxirane can evolve to other final products depending on the additives (Scheme 13).
 | ||
| Scheme 12 Epoxide opening in carvone derivatives. | ||
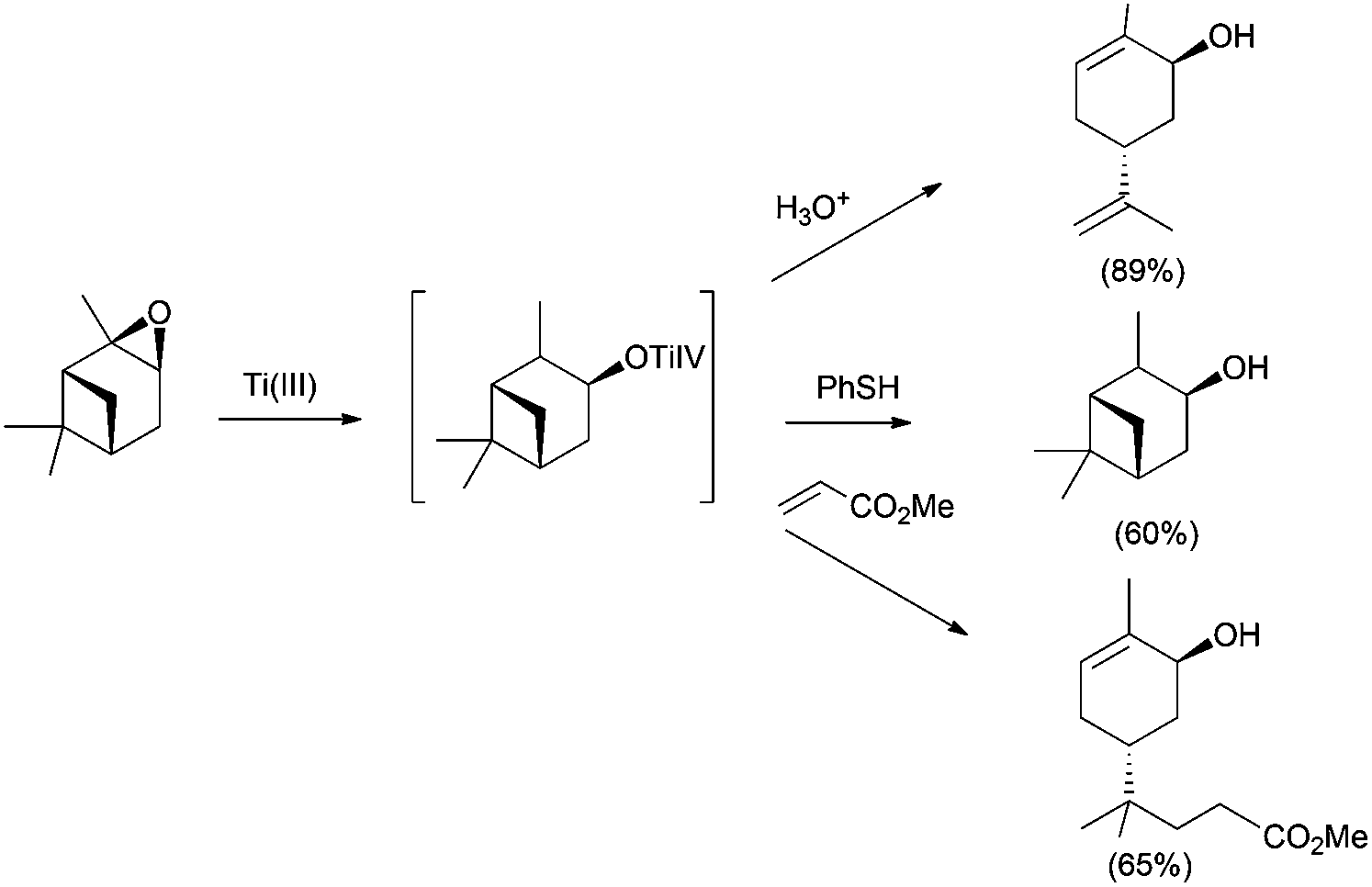 | ||
| Scheme 13 Ti(III)-mediated opening of α-pinene oxide. | ||
The synthesis of one of the most biologically potent and least accessible gibberellins, GA32, was achieved using another Cp2TiCl-mediated epoxide opening reaction as the key step.25 In this case, it was suggested that the initial 1,1-disubstituted epoxide was transformed into an aldehyde by reduction of the radical generated from the oxirane and Cp2TiCl and subsequent hydride elimination to form an alkoxytitanium enolate (Scheme 14).
 | ||
| Scheme 14 Cp2TiCl in gibberellins synthesis. | ||
Reductions
Alternatively to the deoxygenation protocol, radical intermediates, mildly obtained from treatment of an epoxide with Cp2TiCl, are reduced when they are trapped by an H-atom donor, such as 1,4-CHD or t-BuSH (Scheme 15).1,2,26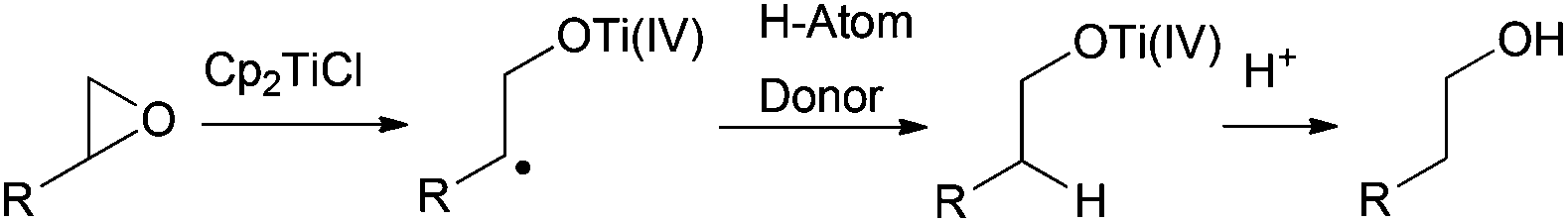 | ||
| Scheme 15 General pathway towards reduced products. | ||
Regioselective ring opening of epoxy alcohols using t-BuSH as the H-atom donor was used in the stereoselective synthesis of α,β-unsaturated-δ-lactones.27 Besides, a chiral 2-methyl-1,3-diol moiety arises from the diastereoselective opening of trisubstituted epoxy alcohols, being the key step in the synthesis of (+)-prelactone C (Scheme 16).28 In this case, 1,4-cyclohexadiene was used as a reducing agent in combination with Cp2TiCl. This efficient synthesis of 1,3-diols from 2,3-epoxy alcohols has been also successfully applied as the key step in the formal synthesis of (+)-antimycin A3b as well as in the total synthesis of (+)-blastmycinone (Fig. 1).29
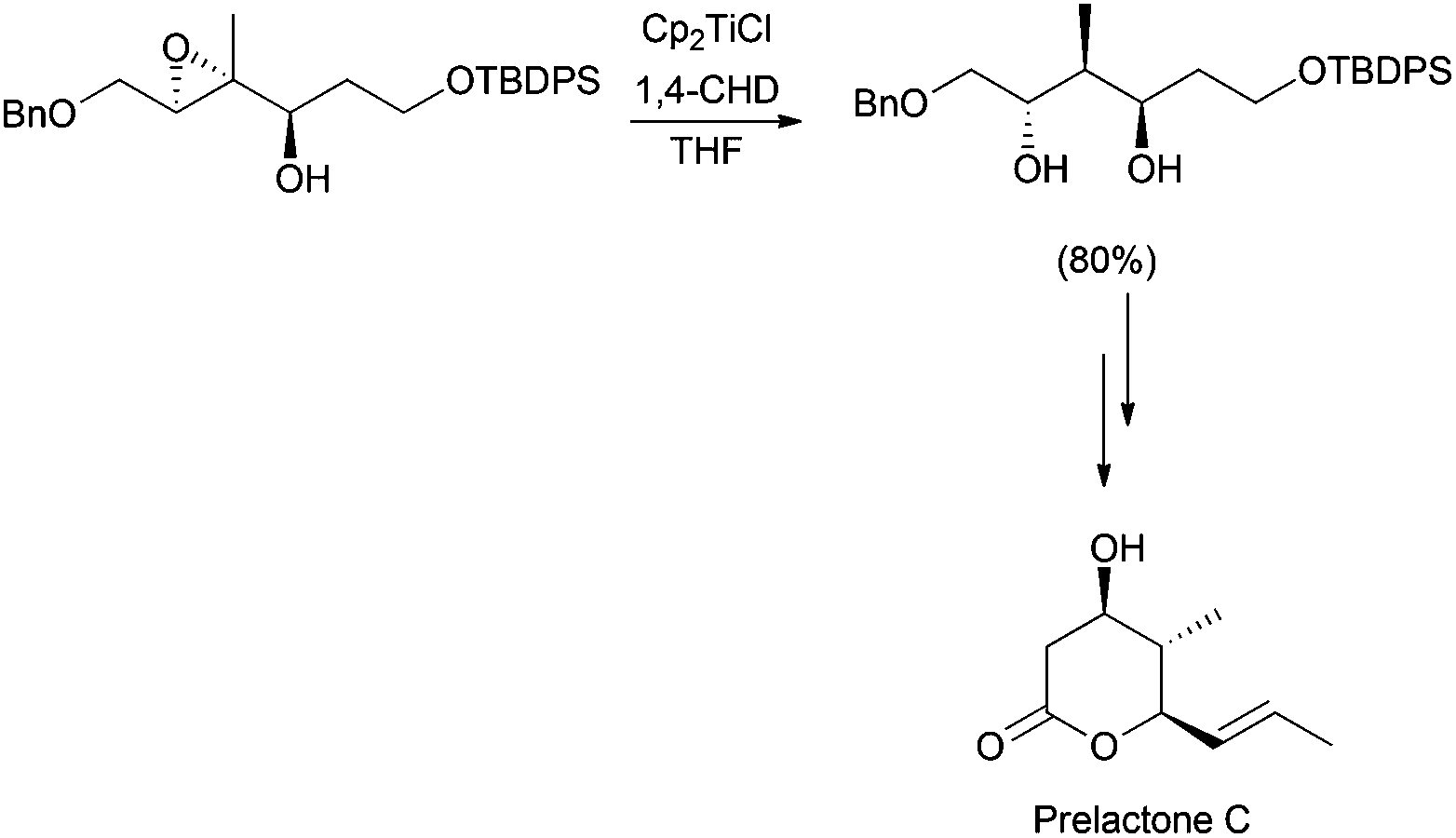 | ||
| Scheme 16 Cp2TiCl in the synthesis of prelactone C. | ||
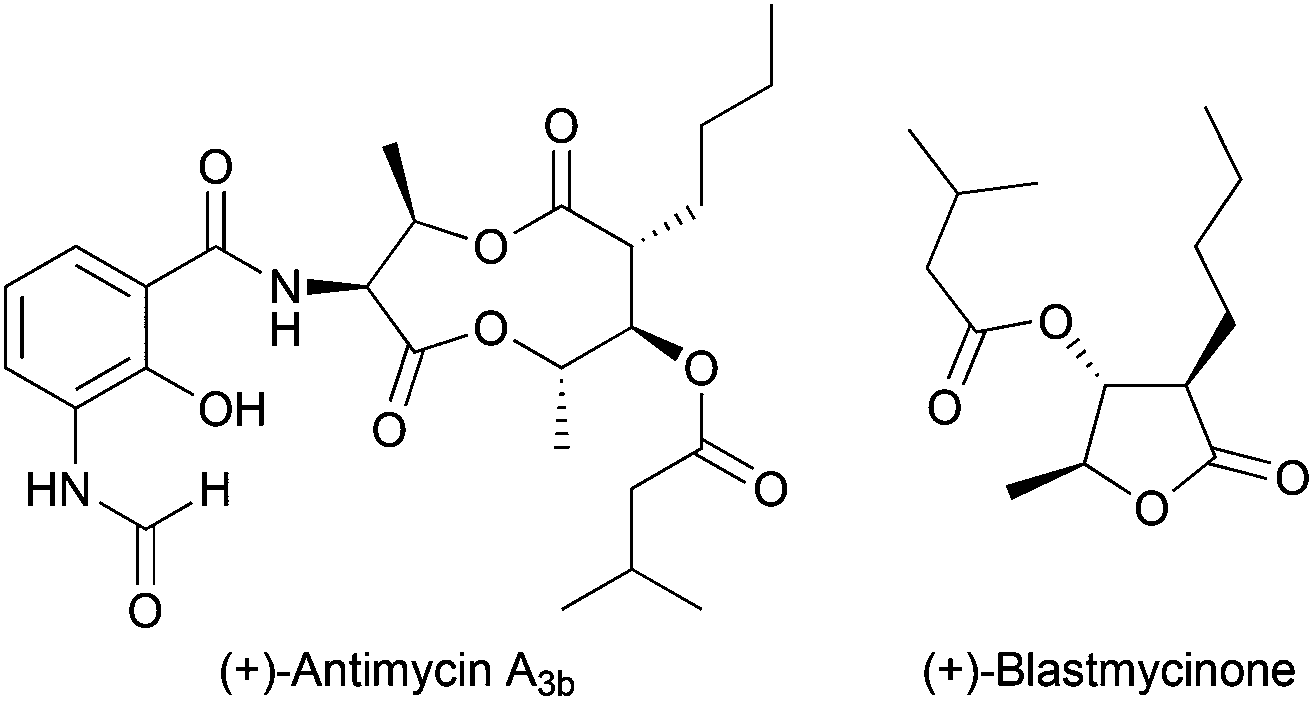 | ||
| Fig. 1 (+)-Antimycin A3b and (+)-blastmycinone. | ||
The regioselective reduction of vinyl epoxides has also been described. Interestingly, E allyl alcohols arise from both E and Z vinyl epoxides 4 (Scheme 17).30 Nevertheless, in the treatment of terminal vinyl epoxides with Cp2TiCl only the deoxygenated product was observed.
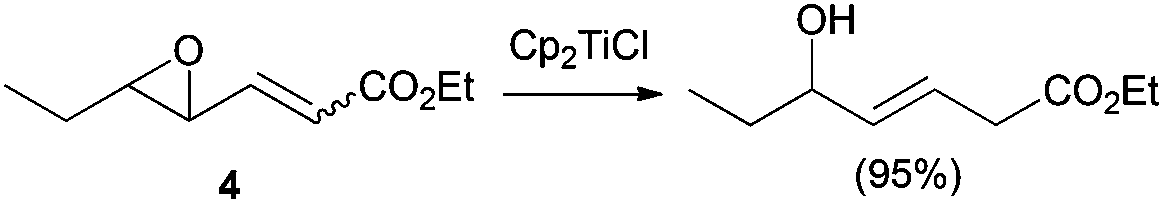 | ||
| Scheme 17 Reduction of vinyl epoxides by Cp2TiCl. | ||
Aldols are significant intermediates in the synthesis of many natural products. A catalytic titanocene-based system has been developed by Doris et al. to achieve the selective reduction of α,β-epoxy ketones to the corresponding β-hydroxy ketones (Scheme 18).31 A year later, this group also reported the selective reduction of α,β-unsaturated ketones to the corresponding saturated ketones mediated by Cp2TiCl.32 This protocol was extended by Kosal and Ashfeld to the catalytic conjugative reduction of α,β-unsaturated aldehydes, ketones, esters and amides.33
 | ||
| Scheme 18 Synthesis of β-hydroxy ketones. | ||
If the generated radicals are trapped intramolecularly, cyclisation reactions can be easily developed. In this sense, the reductive radical cyclisations of epoxy esters have been described using titanocene chloride.34 Thus, for example, chiral quaternary centers can be constructed.35 In this case, the intermediate radical is intramolecularly trapped with an activated alkene (Scheme 19).36
 | ||
| Scheme 19 1,3-Diols from 2,3-epoxy alcohols. | ||
Using similar reaction conditions, the stereospecific synthesis of α-glycosides has been reported from 1,2-anhydro sugars, yielding a free C-2 alcohol, which can be subsequently manipulated (Scheme 20).37 Taking also advantage of this strategy, the synthesis of the pyranopyran ring included in many natural products, such as thyrsiferol, was addressed from C-glycosides (Scheme 21).38
 | ||
| Scheme 20 Preparation of α-glycosides from glycals. | ||
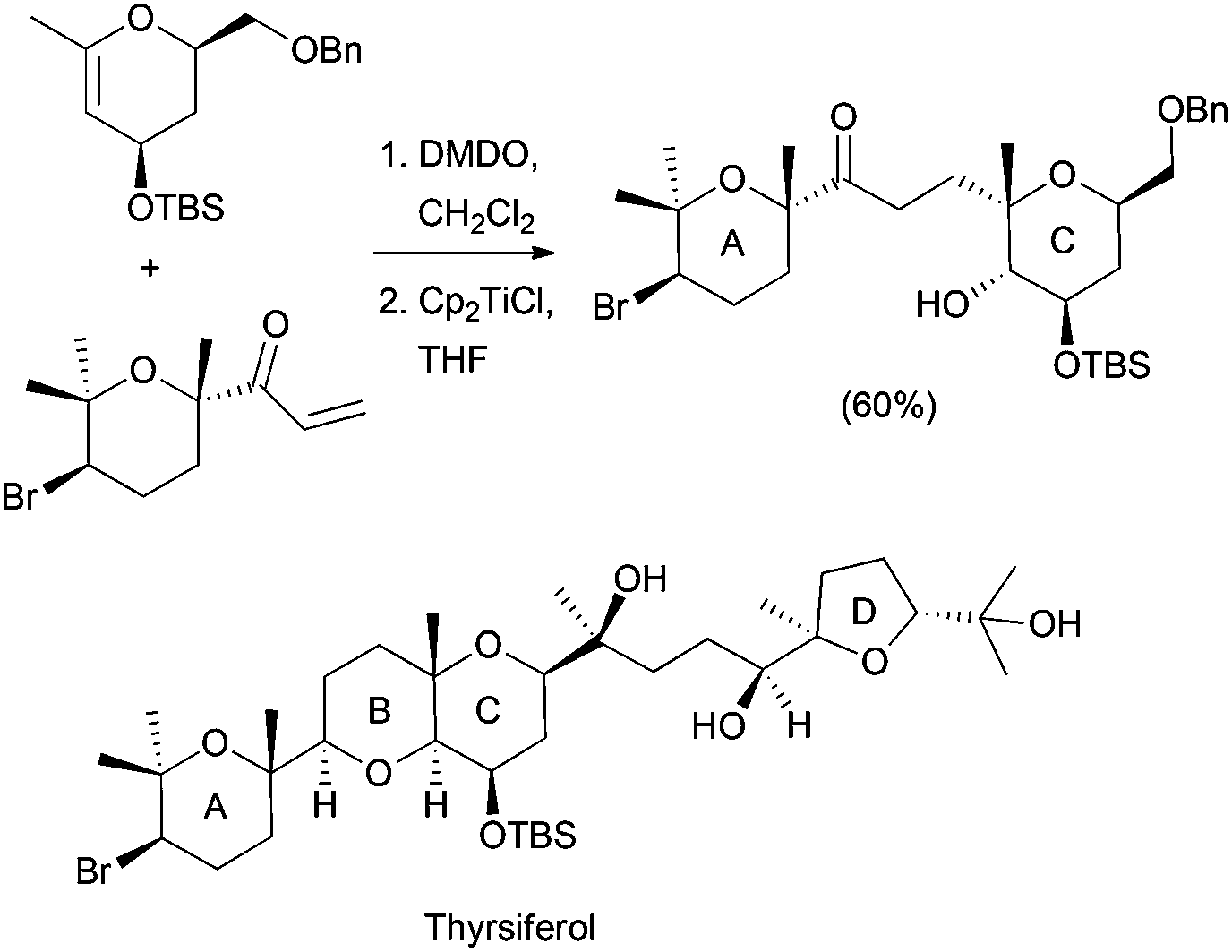 | ||
| Scheme 21 Cp2TiCl in the synthesis of thyrsiferol. | ||
Reductive epoxide ring opening has also been carried out using water as a hydrogen source. Following this procedure, β-deuterated alcohols can be easily prepared in the presence of D2O (Scheme 22).39 Quite interestingly, labelled compounds prepared in this fashion can be applied as an internal standard for analysis, as was shown by using deuterated tyrosol in food analysis.
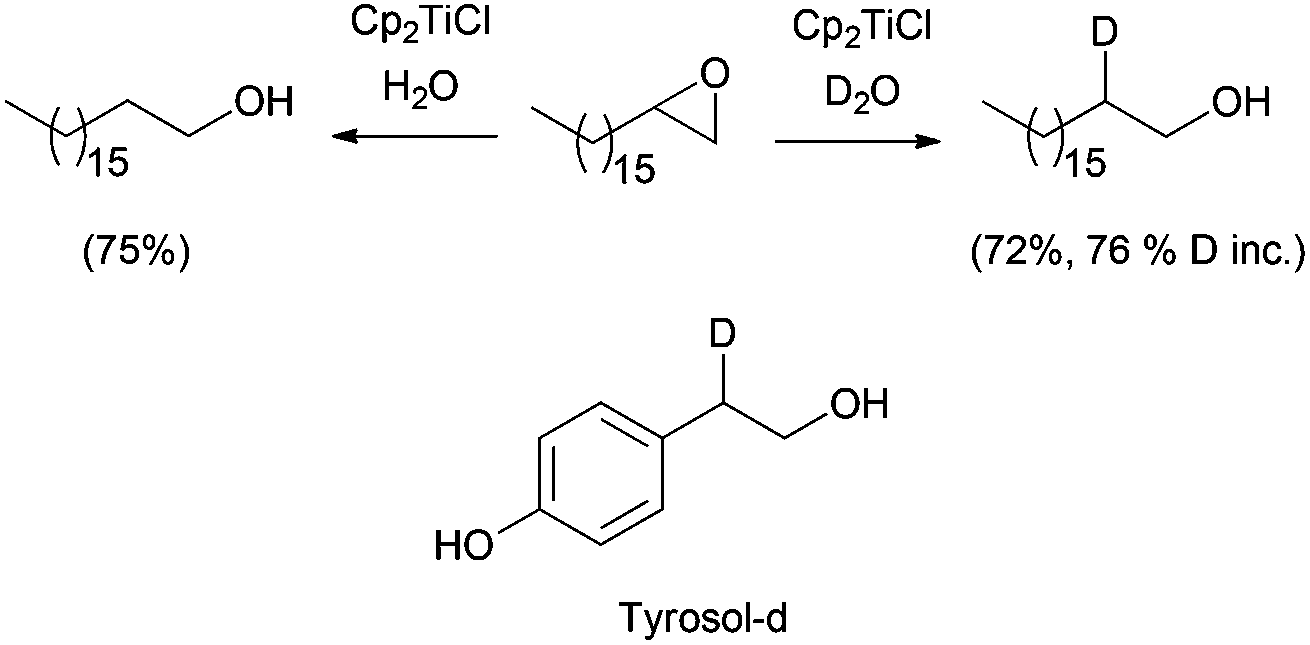 | ||
| Scheme 22 Reductive epoxide opening with Cp2TiCl–water. | ||
A highly regioselective epoxide reduction in the presence of water has been appropriately used in the total synthesis of microcin SF608 (Scheme 23).40
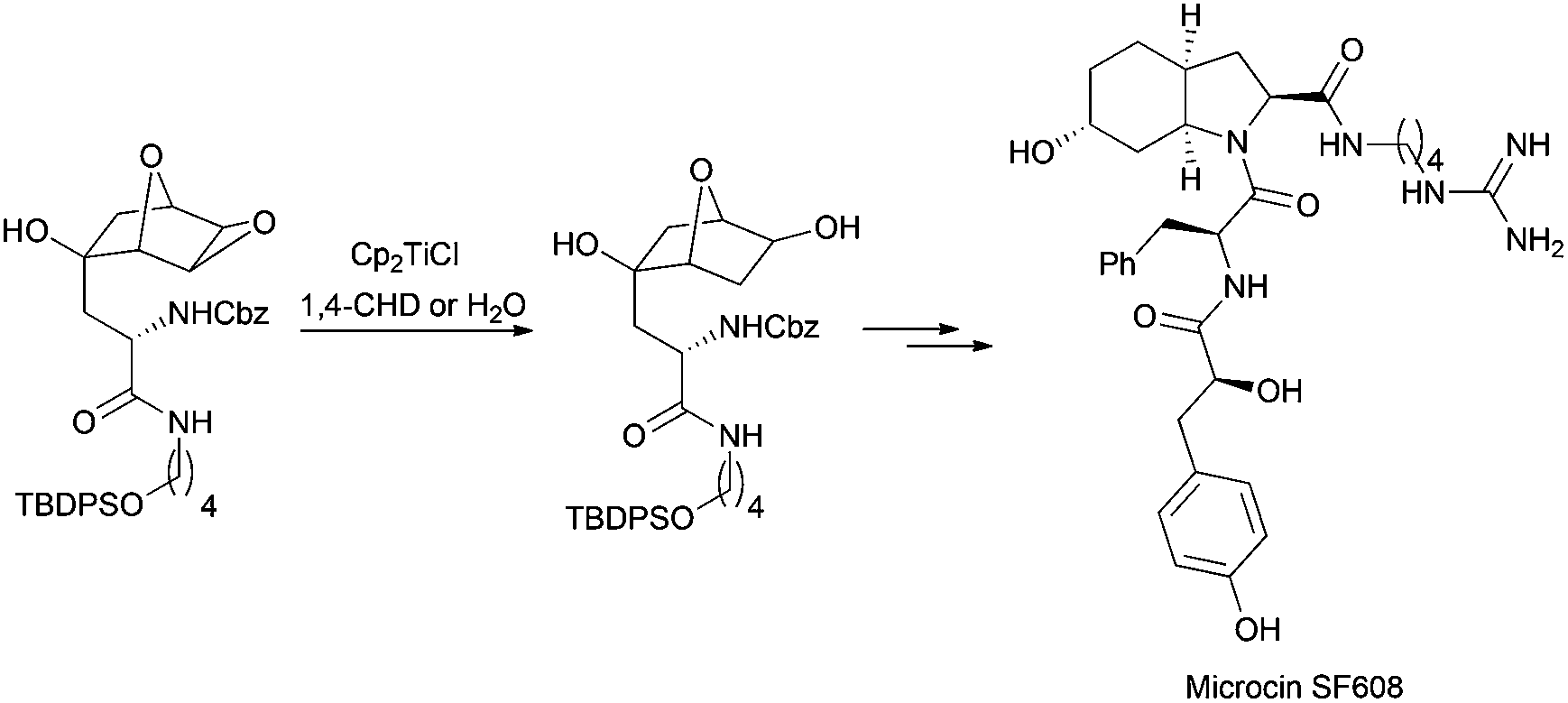 | ||
| Scheme 23 Cp2TiCl in the synthesis of microcin SF608. | ||
Miscellaneous
The group of Roy observed a Cp2TiCl-promoted radical-based Wagner–Meerwein-type rearrangement in camphoric systems, furnishing homoallylic alcohols with good yields (Scheme 24).41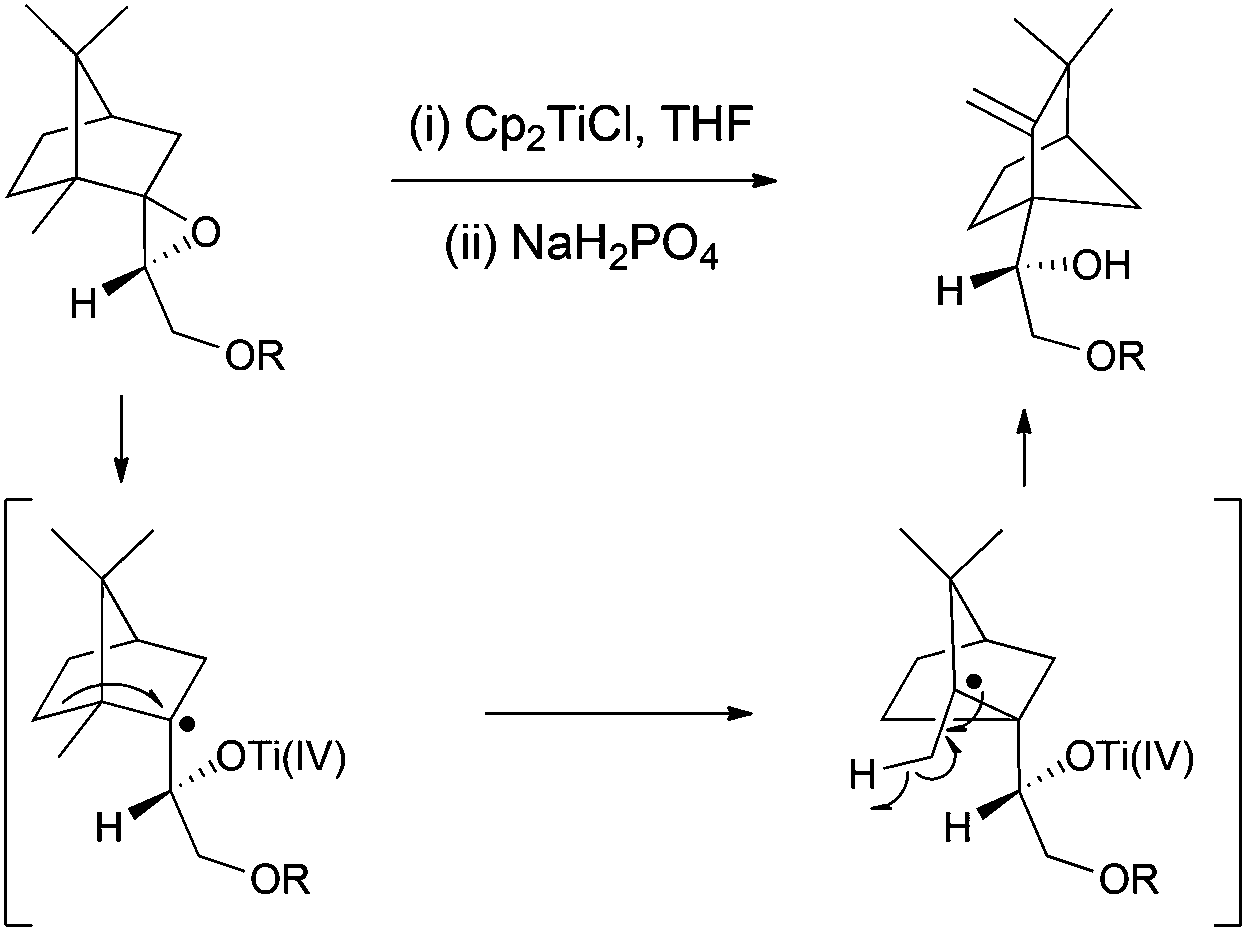 | ||
| Scheme 24 Cp2TiCl-promoted radical-based Wagner–Meerwein-type rearrangement. | ||
Recently, this group has also reported a Cp2TiCl mediated allylation of aldimines for the preparation of homoallyl amines. This methodology has been applied to the formal synthesis of aza-sugar derivatives and alkaloid skeletons (Scheme 25).42
 | ||
| Scheme 25 Cp2TiCl-mediated allylation of aldimines. | ||
Natural product synthesis based on key Cp2TiCl-mediated transformations
Despite the interest of Cp2TiCl in functional group interconversions, one of the remarkable characteristics of this reagent is its capability to build very efficiently different carbon skeletons. This fact has been used as a key step in many natural product syntheses, especially in lignan, terpene and meroterpene synthesis.Lignans
Lignans are a broad and diverse group of compounds, which mainly derive from oxidative couplings of functionalised propyl phenols. These compounds are a major class of phytoestrogens found in plants. Due to their interesting biological properties, such as anticancer and antioxidant activities, lignans have attracted the attention of organic chemists.The group of Roy has developed a reliable methodology for the synthesis of furano and related lignans, based on free radical chemistry mediated by in situ generated Cp2TiCl from Cp2TiCl2 and zinc dust. Cp2TiCl has therefore been used for the synthesis of the key tetrahydrofuran ring. The radical cascade process consists of a Cp2TiCl-mediated homolytic epoxide opening (Scheme 3), an intramolecular radical cyclisation followed by an iodine-mediated etherification reaction (Scheme 26).43
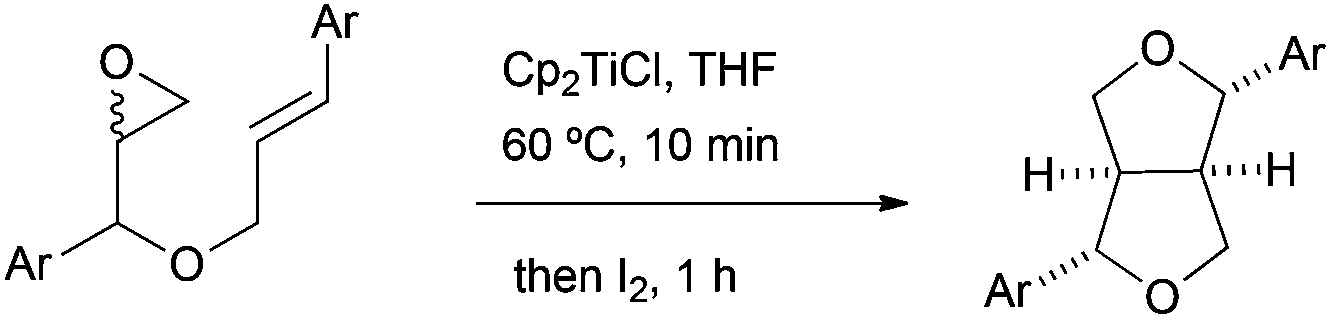 | ||
| Scheme 26 Intramolecular radical cyclisation and etherification. | ||
This protocol has been used by the authors for the synthesis of the following furano lignans: dihydrosesamin 5a,44 lariciresinol dimethyl ether 5b, acuminatin methyl ether 5e, sanshodiol methyl ether 5g, lariciresinol 5h, acuminatin 5i, and lariciresinol monomethyl ether 5j. A similar approach yielded different furofuran lignans, such as sesamin 6a, eudesmin 6b, piperitol methyl ether 6e, pinoresinol 6h, piperitol 6i, and pinoresinol monomethyl ether 6j,45 (Fig. 2).
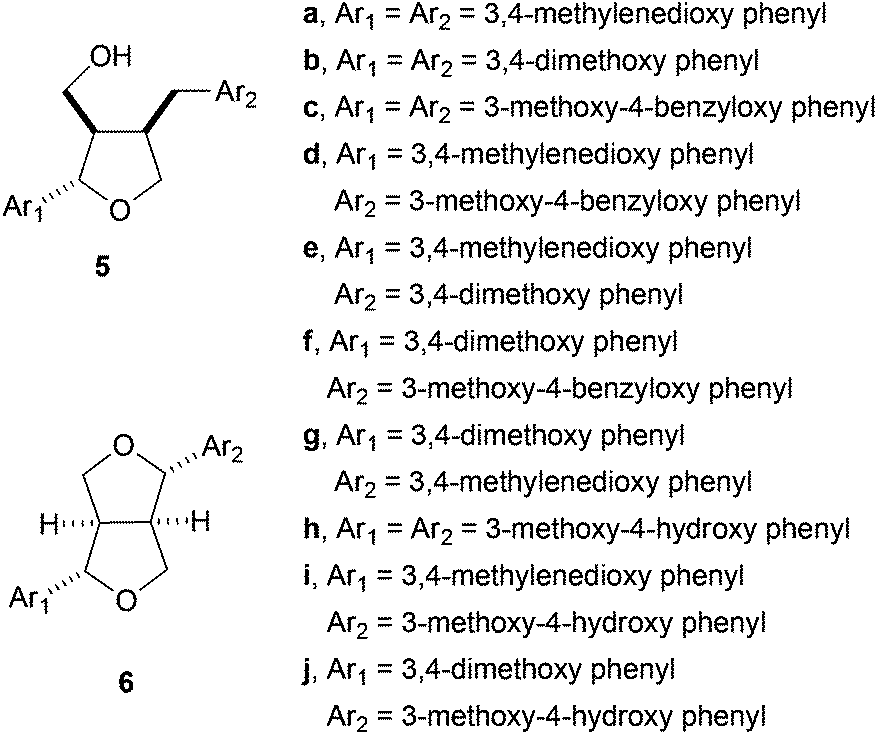 | ||
| Fig. 2 Furan- and furofuran lignans prepared using Cp2TiCl. | ||
Lately, the same group achieved the enantioselective synthesis of (−)-dihydrosesamin, (−)-acuminatin, (−)-sesamin and (−)-methyl piperitol starting from the corresponding chiral epoxy alcohol prepared by the Sharpless kinetic resolution method46 (Scheme 27).
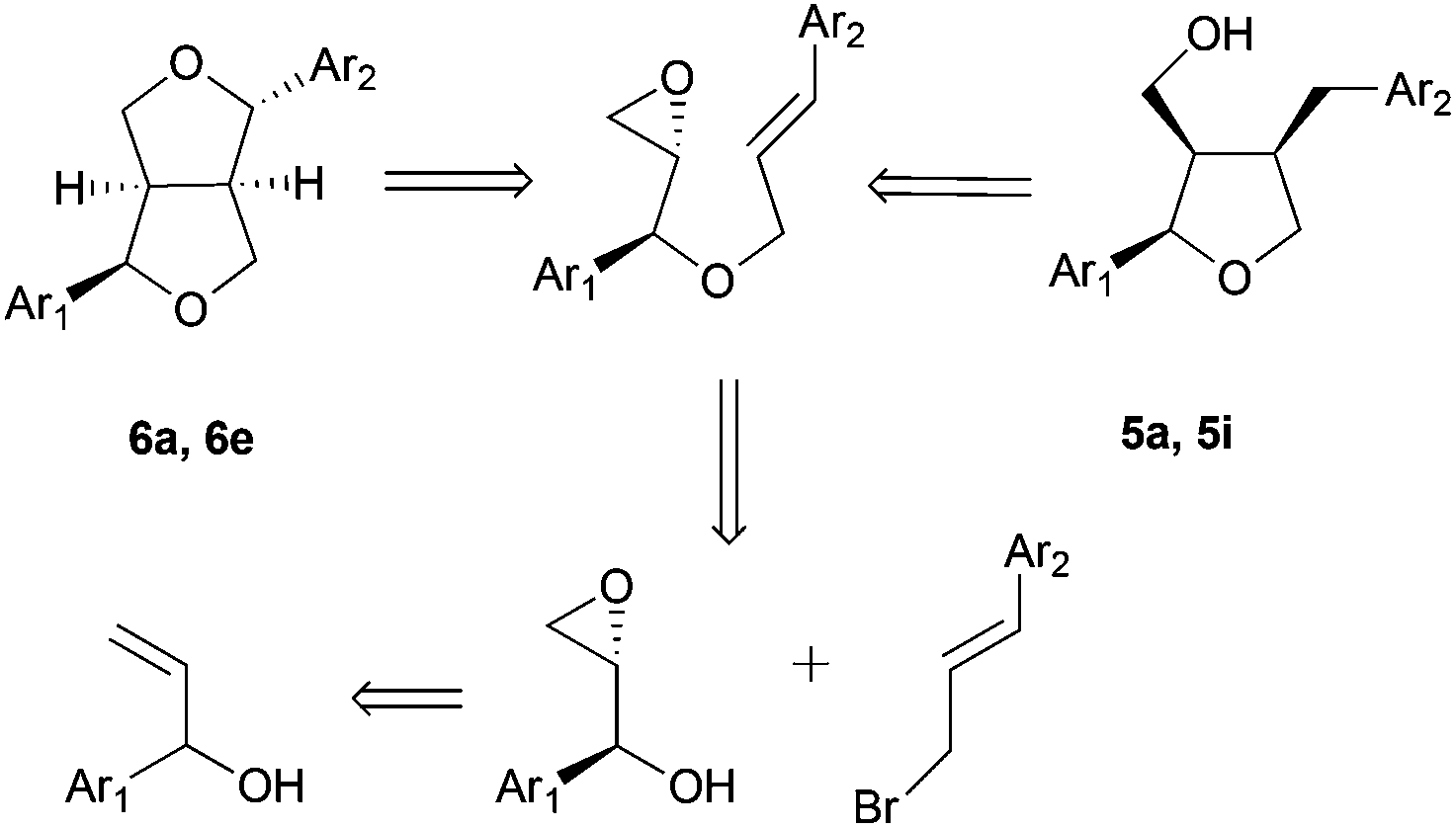 | ||
| Scheme 27 Retrosynthetic analysis. | ||
Recently, the same group has reported the formal synthesis of the furano lignans magnofargesin 7 and 7′-epimagnofargesin 8 in both racemic and optically pure forms through a similar protocol.47 In this case, the Cp2TiCl-mediated radical cyclisation reaction was conducted from the epoxyalkyne 9 to give the tetrahydrofuran ring with an external alkene, which is characteristic of these compounds (Scheme 28).
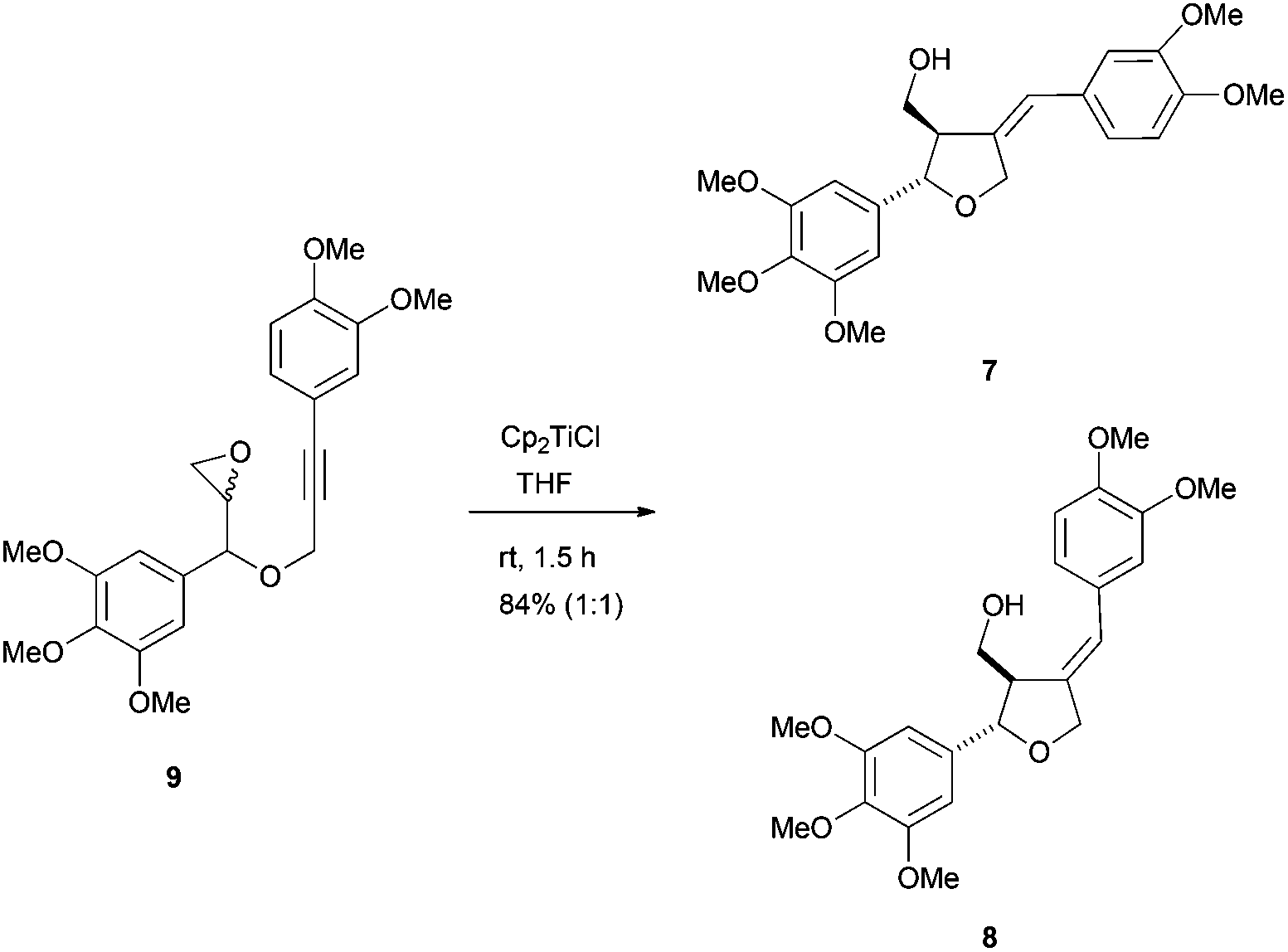 | ||
| Scheme 28 Cp2TiCl mediated the intramolecular radical cyclisation of the epoxyalkyne 9. | ||
Terpenes
The use of Cp2TiCl-mediated cyclisations constitutes a straightforward strategy for the synthesis of natural terpenes, and therefore has been extensively used especially during the last 10 years.48 As mentioned above, this reagent is able to promote regioselective homolytic ring opening of epoxides (Scheme 3). In the context of terpene synthesis, the radical thus generated is able to give multiple intramolecular additions to alkenes and alkynes, generating the corresponding cyclic products (see Scheme 29).1 Remarkably, Cp2TiCl can now be used in substoichiometric amounts with the aid of different Ti(III)-regenerating agents. This fact has contributed to the development of enantioselective processes and also to control the concentration of the active species in the reaction medium.11,49 The latter fact is very important to avoid premature trapping of intermediate radicals during a (poly)cyclisation process. Moreover, in many cases, Cp2TiCl is able to oxidise the final radical yielding an alkene, thus mimicking a pure cationic process. In this sense, if the global process is considered, including epoxide opening, stereoselective (poly)cyclisation and the oxidative ending, this complex could be assumed to be an efficient artificial cyclase.50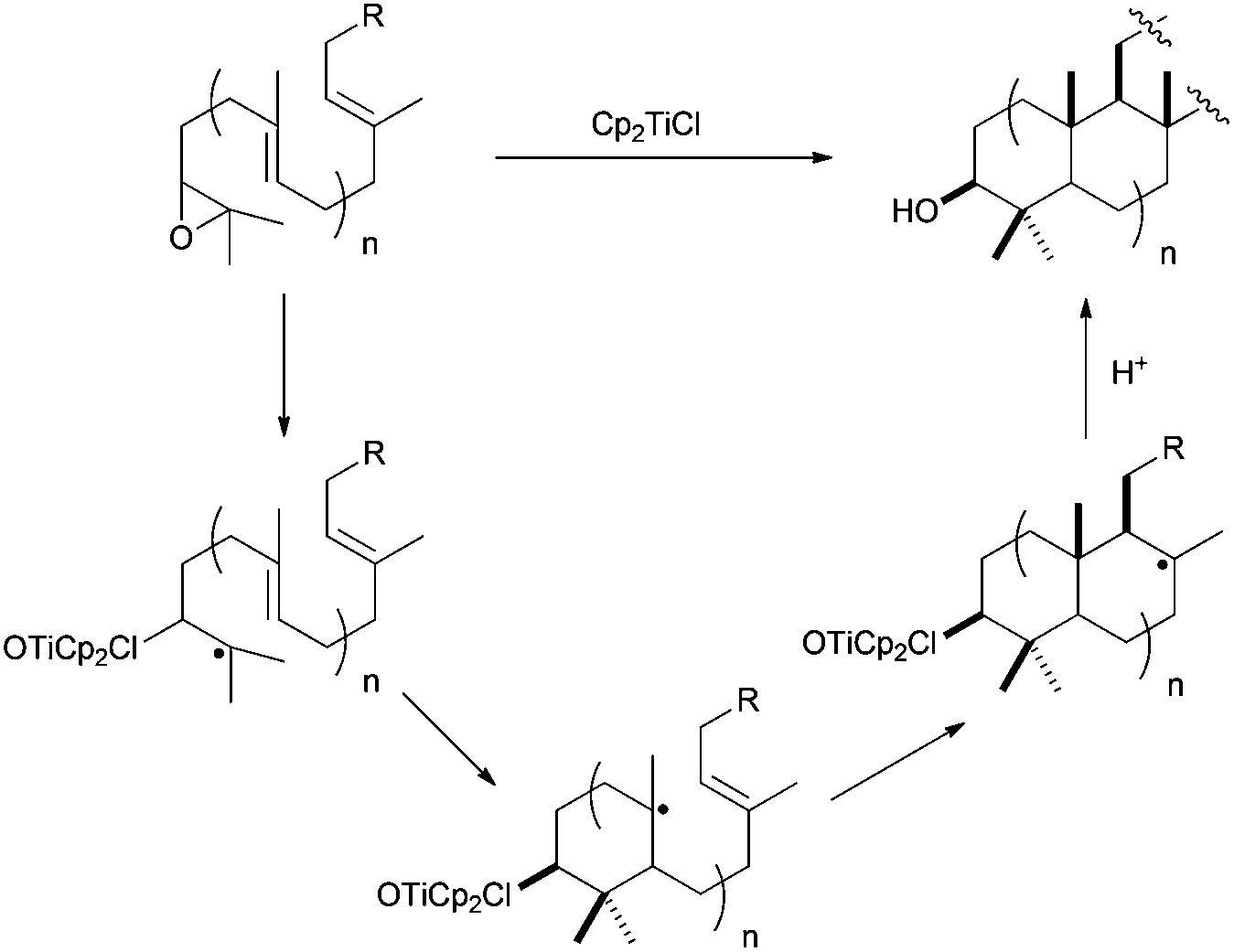 | ||
| Scheme 29 Example of Cp2TiCl mediated epoxypolyprene cyclisation. | ||
Historically, Clive et al. reported in 1995 the first example of a Cp2TiCl-mediated cyclisation as the key step for the synthesis of a natural sesquiterpene, ceratopicanol (Scheme 30). In this case, the last cyclopentane ring is prepared by a 5-exo-dig monocyclisation reaction in good yield.51a,b A similar strategy has been recently used in the preparation of sesquiterpenes merrilactone A and anislactone A.51c
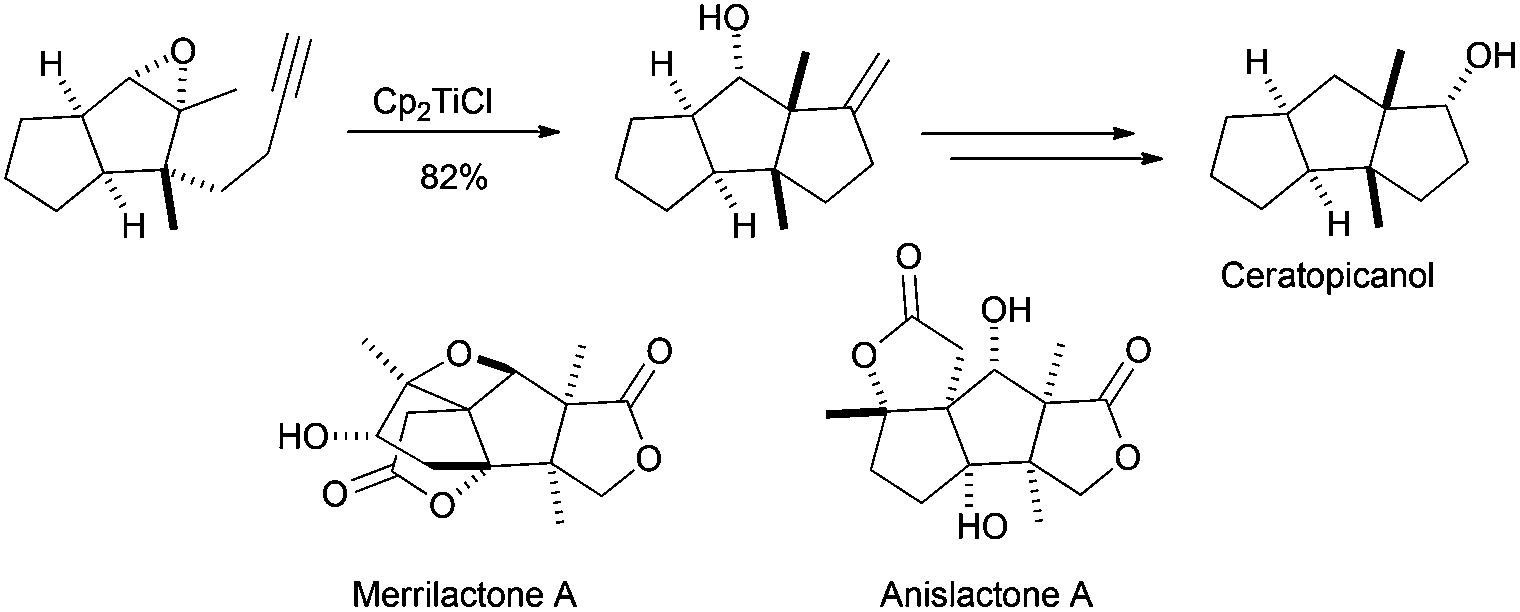 | ||
| Scheme 30 Synthesis of ceratopicanol using a Cp2TiCl-mediated 5-exo-dig cyclisation. | ||
The development of a more general Cp2TiCl-based approach to conventional terpenic skeletons was reported by Cuerva et al. in 2001.52 In that work, the authors presented the synthesis of several mono and bicyclic terpenic frameworks using stoichiometric amounts of Cp2TiCl from simple epoxypolyprenes. The yields were moderate to good, with excellent stereoselectivities, mainly yielding cyclisation products with exo double bonds. Nevertheless, the control of the final oxidative ending was not good, also giving the corresponding endo double bonds and reduction products (Scheme 31). The formation of an exo double bond was lately assigned to a mixed disproportionation reaction between the organic radicals and Cp2TiCl.13 On the other hand, the presence of reduced products was due to a very efficient hydrogen atom transfer (HAT) process from a titanocene(III) aquocomplex, formed by the presence of adventitious water, to the corresponding carbon-centred radical.9,53
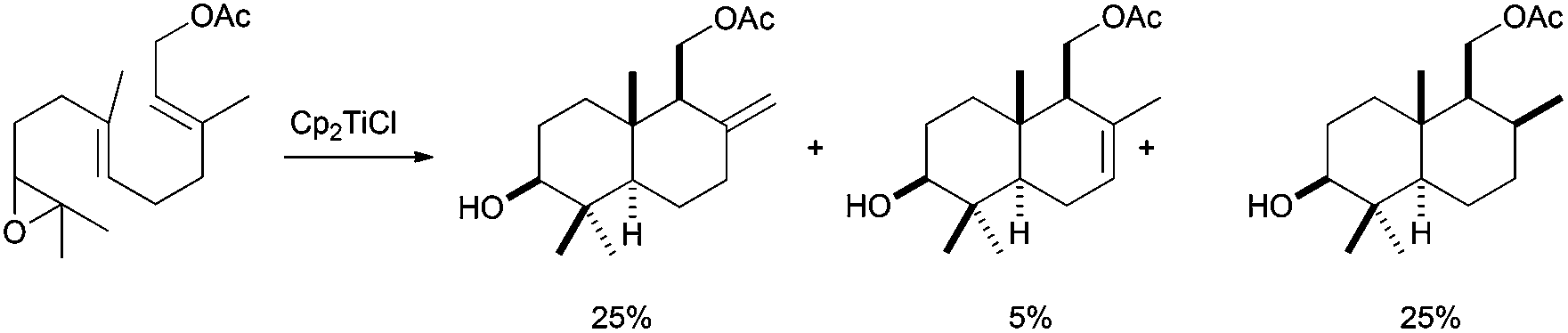 | ||
| Scheme 31 First example of a Cp2TiCl-mediated epoxypolyene cyclisation. | ||
Similar results were obtained by Takahashi's group in an approximation to the synthesis of taxol based on the monocyclisation of epoxygeranyl acetate and other closely related functionalised derivatives.54
The methodology developed by Cuerva et al. was used to confirm the structure of monocyclic triterpene achilleol A (8),55 a metabolite from Achillea odorata,56 taking advantage of the excellent characteristics of this kind of radical cyclisation to generate exocyclic double bonds (Scheme 32). In this case, the key intermediate is monocycle 10, which presents the required exocyclic double bond and derives from the corresponding epoxide of protected geranylacetone. This monocyclic compound was transformed into achilleol A (12) in a few steps. This cyclic synthon 10 was subsequently used in the synthesis of the cyclofarnesane terpenoid 1111 and achilleol B (13).57
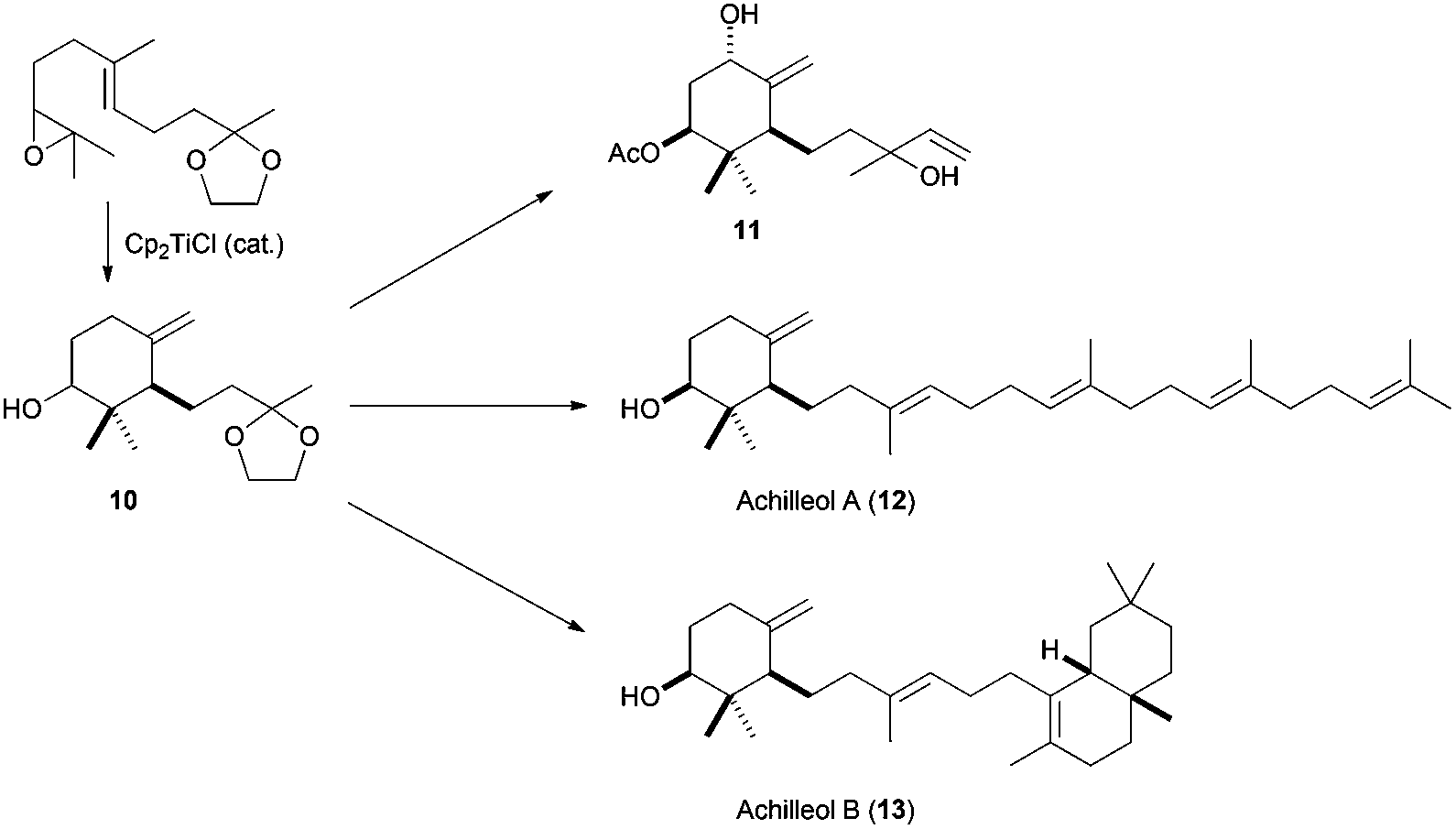 | ||
| Scheme 32 Synthesis of achilleol A, B, and 11 from products obtained by Cp2TiCl-catalysed epoxyolefin cyclisation. | ||
Additionally, other bicyclic terpenes, such as labdane 3β-hydroxymanool,11 isolated from Gleichenia japonica, or sclareol oxide,58 were synthesised by similar procedures, but using the corresponding cyclisation product derived from the epoxide of protected farnesylacetone (Scheme 33). Recently, Álvarez-Manzaneda et al. have also used compound 14 in the total synthesis of negundoin A.58b
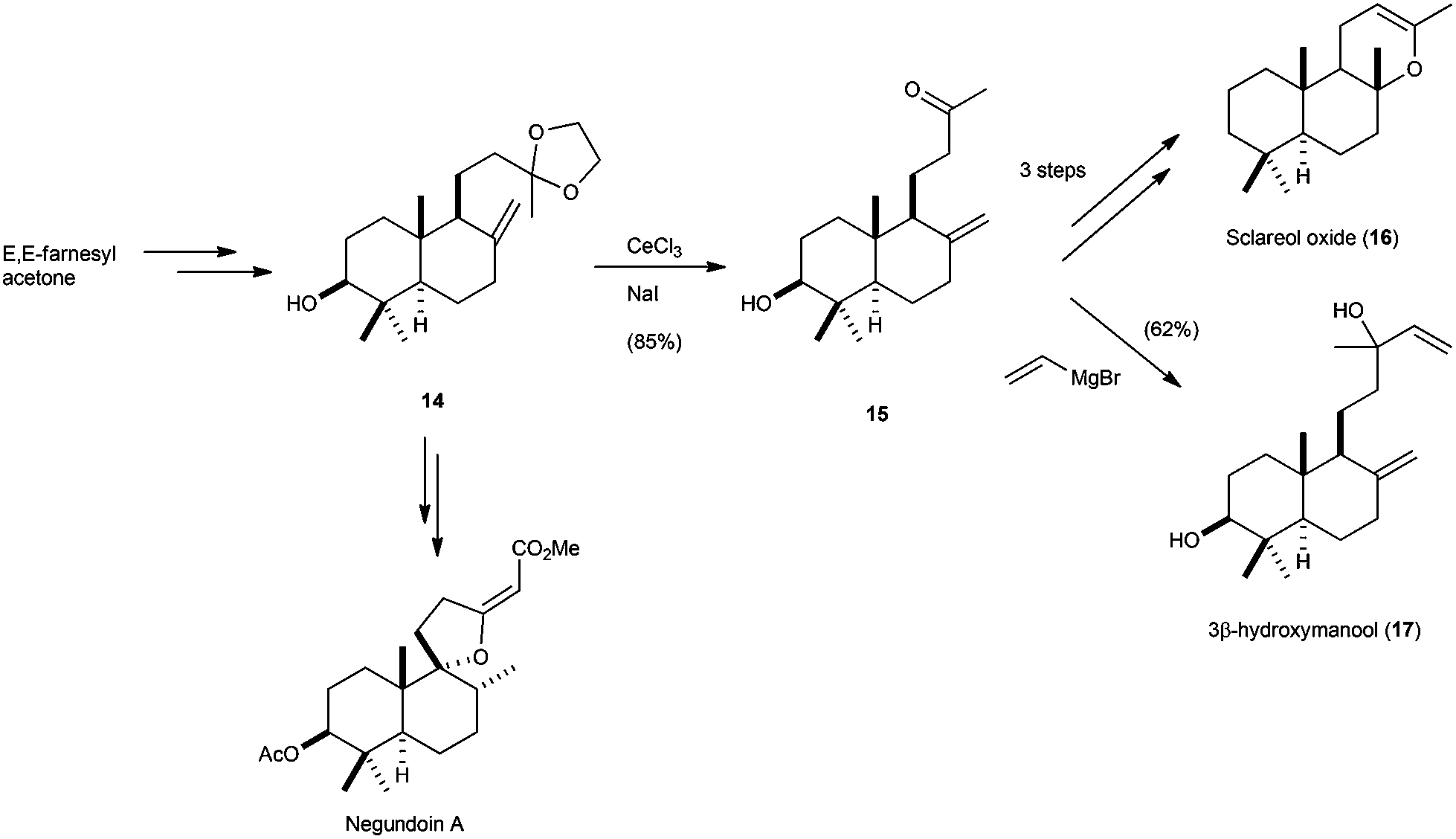 | ||
| Scheme 33 Synthesis of sclareol oxide, 3β-hydroxymanool, and negundoin A via Cp2TiCl-catalysed epoxypolyprene cyclisation. | ||
Synthesis of natural terpenes based on higher polycyclisations has been also described, such as furanoditerpenoids 20 and 21, prepared from Cp2TiCl-catalysed cyclisation of geranylgeranyl acetate previously described by Cuerva et al.,11 to yield the tricyclic compound 19, which could be transformed into the furanoditerpenoids 20 and 21 in a few steps (Scheme 34).59
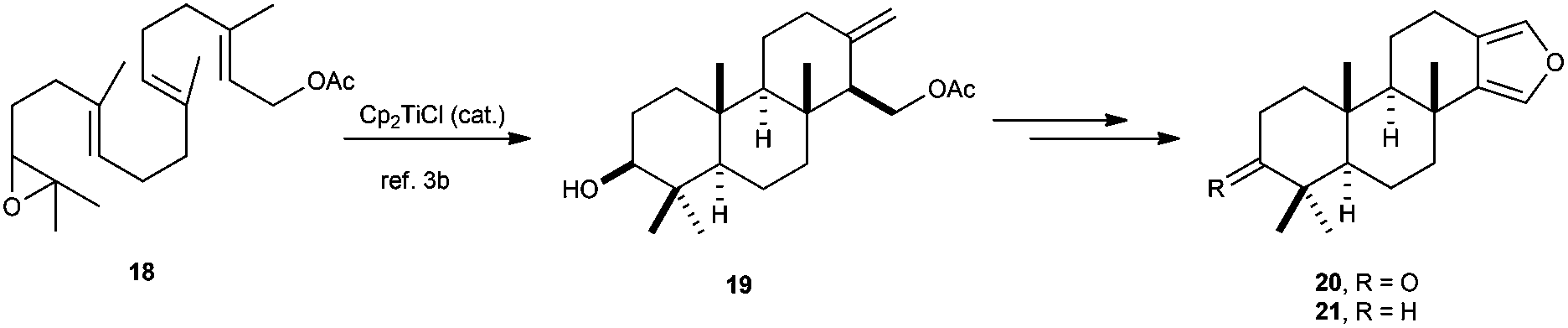 | ||
| Scheme 34 Synthesis of furanoditerpenoids 20 and 21via Cp2TiCl-catalysed cyclisation of 18. | ||
The versatility as building blocks in terpene synthesis of compounds generated by titanocene(III)-catalysed cyclisations of simple epoxypolyprenes is also revealed in the preparation of several bicyclic compounds from the family of drimanes. Taking advantage of the exocyclic double bond in drimanic skeleton 22, Cuerva et al. prepared a collection of 3β-hydroxydrimanes with different functionalisations, as isodrimenediol (23), drimane (24), 3β-hydroxydihydroconfertifolin (25), 3β-hydroxycinnamolide (26), and 3β-acetoxydrimenin (27)(Scheme 35).60
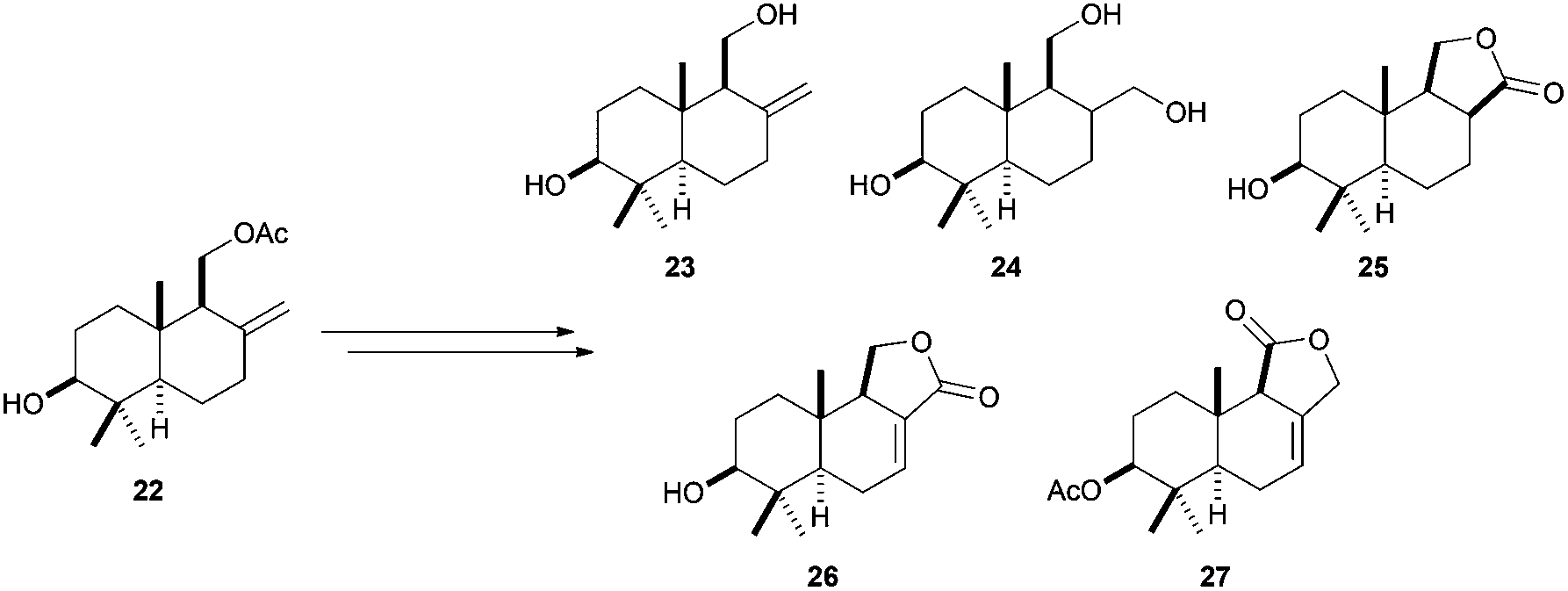 | ||
| Scheme 35 Synthesis of 3β-drimanes from cyclization product 22. | ||
Besides the hydroxyl group at C-3, it is very common in nature to find terpenes with additional oxygenated functions in different parts of the skeleton.61 A frequent arrangement is a γ-dioxygenated system on the A ring. The introduction of this hydroxyl group in non-functionalised cyclisation products is not trivial, and procedures based on allylic oxidations, remote functionalisations or microbiological oxidations normally give low yields or mixtures of compounds, also presenting an important lack of chemoselectivity.62 To overcome these problems, some approximations have been developed. Thus, a two-step protocol consisting of allylic oxidation of the methyl group in terminal alkene of the starting polyprene followed by an epoxidation yields the corresponding hydroxylated epoxypolyprenes. After protection of the hydroxyl group, these compounds were cyclised using Cp2TiCl, generating the corresponding cyclisation products with a γ-dioxygenated system on the A ring, but normally with low yields and moderate stereoselectivities. This strategy has been used by several authors in the preparation of different compounds. In this sense, the synthesis of the taxol-related compound baccatin III (28) was accomplished using a γ-dioxygenated system derived from geranyl acetate (Scheme 36).63 In the same way, mechanistic studies about the cyclisation of these functionalised epoxypolyprenes have been performed.64
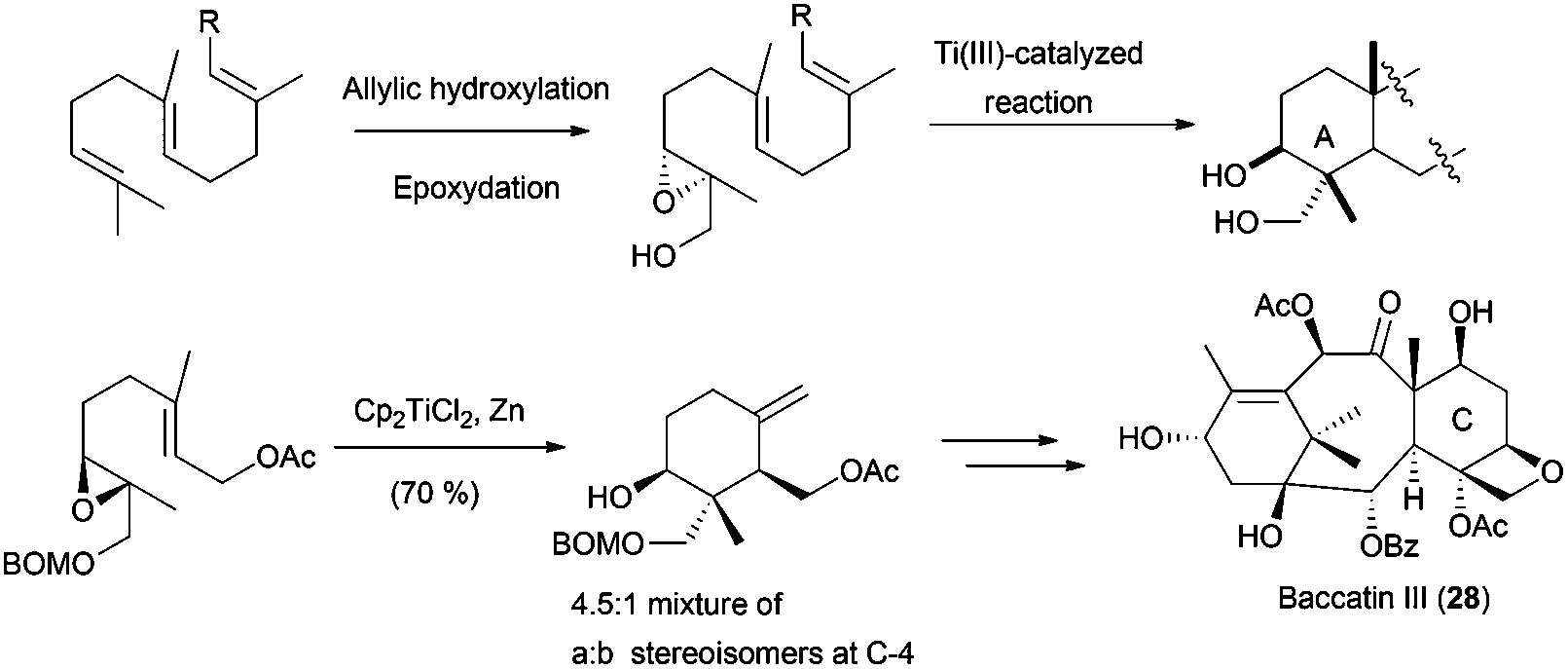 | ||
| Scheme 36 Hydroxyl-promoted epoxidation strategy and application to natural products synthesis. | ||
An alternative to this methodology for the synthesis of the γ-dioxygenated system on the A ring was developed by Cuerva et al., using the hydroxyl group located at C-3 as a template to achieve a remote C–H activation.65 The placement at C-3 of an oxime and subsequent treatment with Na2PdCl4 allows the formation of a five-membered palladium complex, involving the oxime and the equatorial methyl group at C-4. The final oxidation of this complex generated regioselectively the introduction of an acetoxy group at that position (Scheme 37).
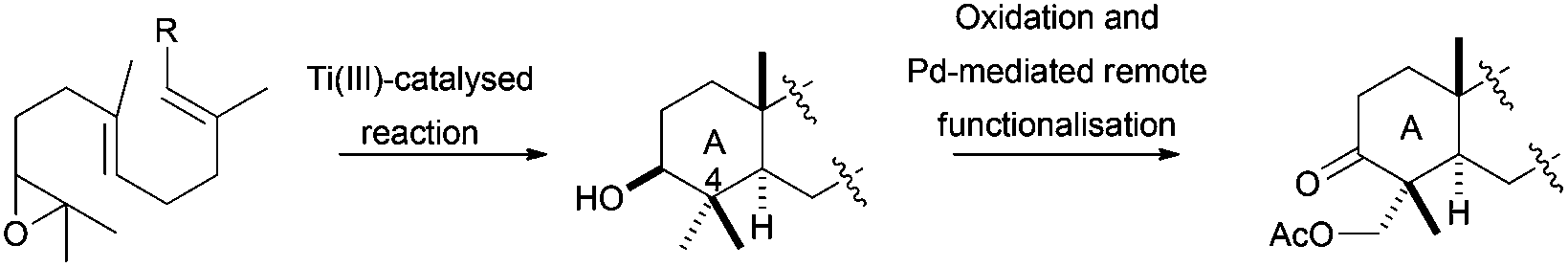 | ||
| Scheme 37 Post cyclisation oxidation of products obtained via Cp2TiCl catalysed epoxypolyene cyclisation. | ||
Using this procedure, the authors carried out the functionalisation of several mono-, bi- and even tricyclic terpenic skeletons. Some of these compounds were used in the short and efficient synthesis of natural terpenoids. Thus, rostratone, isolated from N. rostrata,65 was prepared from cyclisation compound 29, which was transformed in 30 using simple transformations (see Scheme 38). Finally, the bicyclic compound was functionalised using the Pd-based methodology and a subsequent transesterification yield rostratone (32). Additionally, formal total syntheses of complex natural terpenes aphidicoline and pyripyropene A were also accomplished.
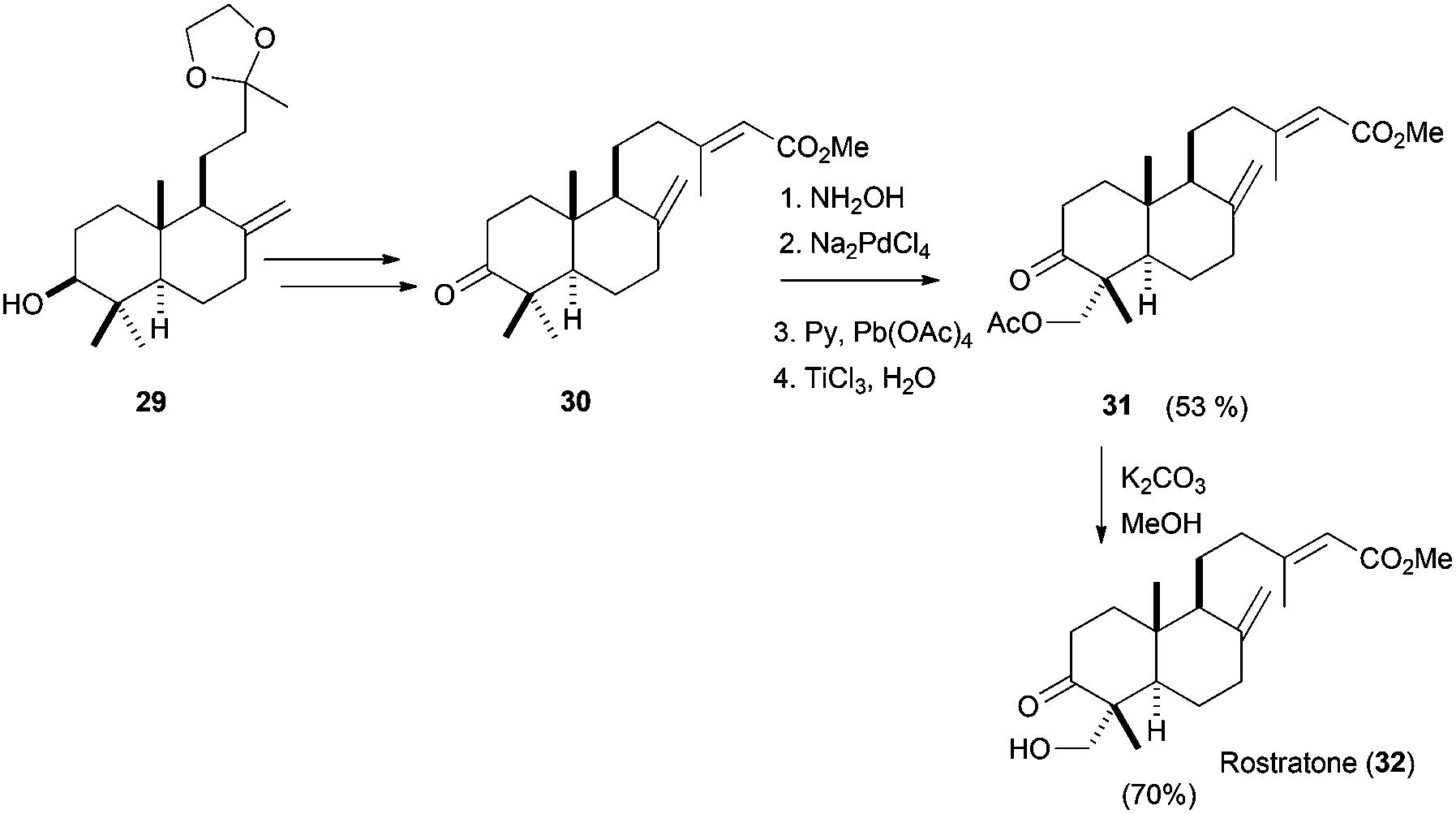 | ||
| Scheme 38 Synthesis of rostratone from epoxypolyene cyclisation product 29. | ||
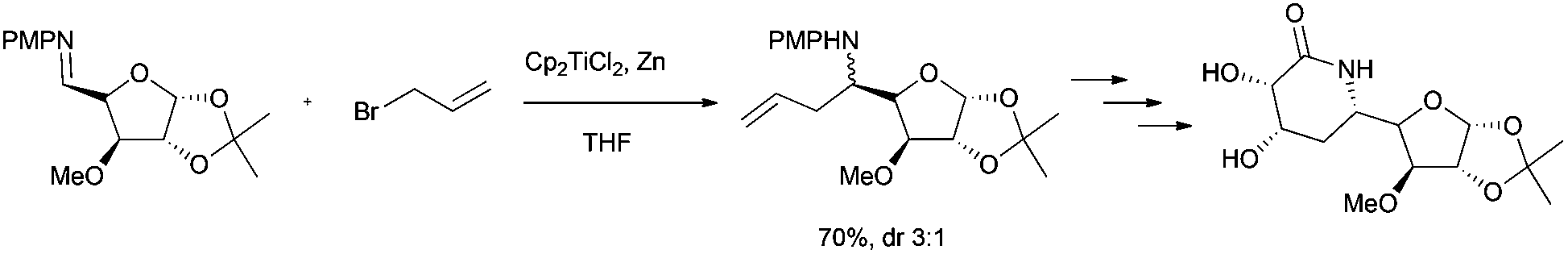
Bioinspired Cp2TiCl-promoted processes have also allowed the synthesis of several terpenic structures hydroxylated in different positions of the terpenic frameworks, such as C-1, C-7 or C-12. These polyhydroxylated structures are also very common in natural terpenes. In this context, Cuerva's group has applied the versatile chemistry of titanocene(III) complexes to the development of a new methodology to gain access to such polyhydroxylated structures.66a It consists of an initial Cp2TiCl-promoted Barbier reaction of appropriate α,β-unsaturated aldehydes with simple allyl bromides (as prenyl, geranyl or farnesyl bromides) to generate the corresponding hydroxylated polyprenes (Scheme 4). Then, after subsequent protection of the hydroxyl group and regioselective epoxidation, a Cp2TiCl-catalysed cyclisation of the corresponding polyhydroxylated epoxypolyprenes generates the desired mono-, bi-, tri- and tetracyclic polyhydroxylated terpenes with moderate to good yields. Some of these polycyclic products were used in the synthesis of complex natural terpenes, such as sestertatin 1 (see Scheme 39) and C,D rings of the marine natural product aspergilloxide. Sestertatin 1 and aspergilloxide are tetracyclic sesterterpenoids isolated from the sponge Hyrtios erecta67a and an undescribed member of a fungus from the genus Aspergillus,67b respectively. A similar protocol has been recently used in the preparation of (+)-apotrisporin E and (+)-apotrientriols A–B.66b
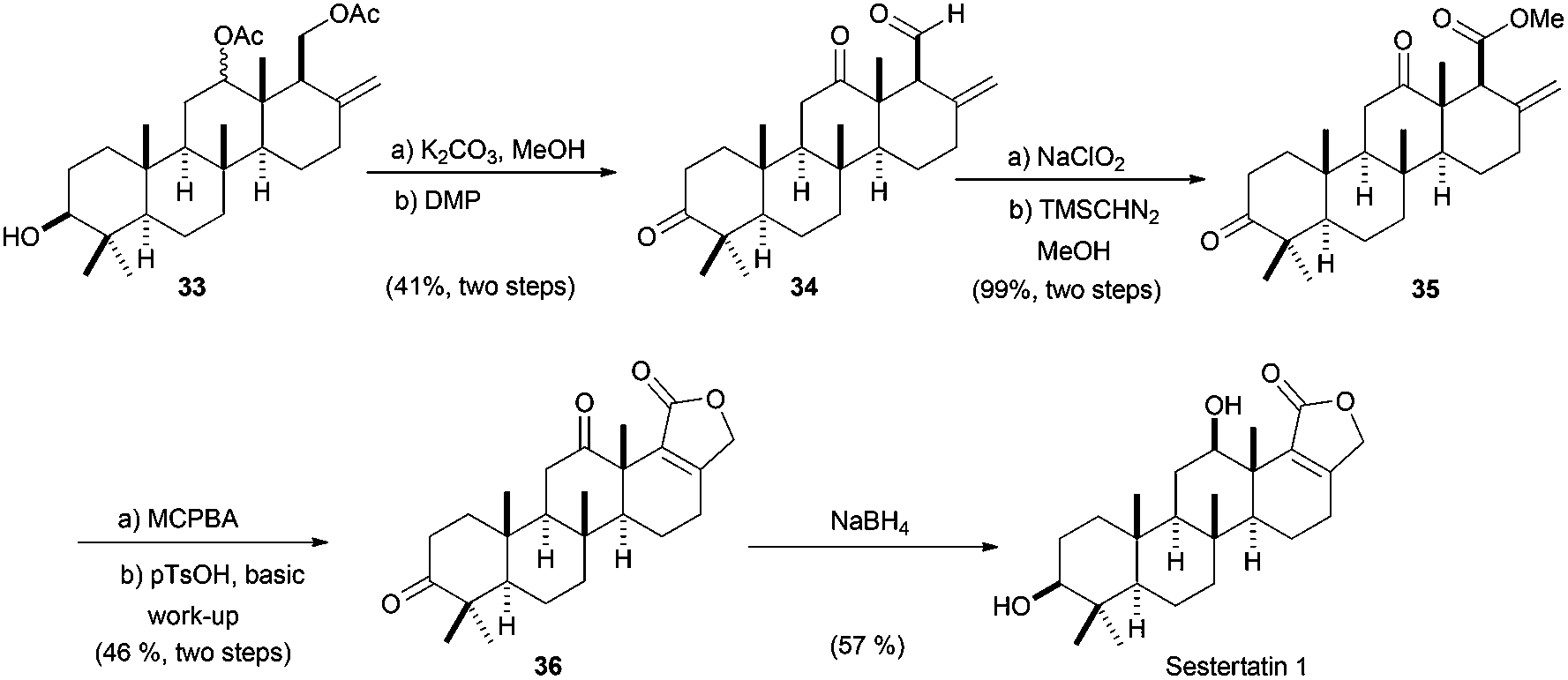 | ||
| Scheme 39 Total synthesis of sestertatin 1 from cyclisation product 33. | ||
Another important aspect in the synthesis of natural products is the development of methodologies which allow the enantioselective access to the desired compounds. In this sense, Cp2TiCl-catalysed bioinspired radical cyclisations of epoxypolyprenes have been shown to be an excellent procedure for the enantioselective synthesis of natural terpenoids. This is because the correct absolute configuration present in the natural terpene can be placed in the starting polyprene by an enantioselective epoxidation process. In this sense, Sharpless dihydroxylation68 has been used for the preparation (after the corresponding epoxide ring closing) of enantioenriched epoxypolyprenes, which retain the enantiomeric excess after the corresponding cyclization. This method has been used in the preparation of several interesting natural terpenes, such as achilleol B (13),57 myrrhanol (39),69 (+)-seco-C-oleanane (38),70 and β-onocerin (37),7 starting from enantioenriched farnesyl epoxide. β-Onocerin was also prepared using a Cp2TiCl-catalysed Wurtz-type dimerisation of allylic halides (Scheme 40).71
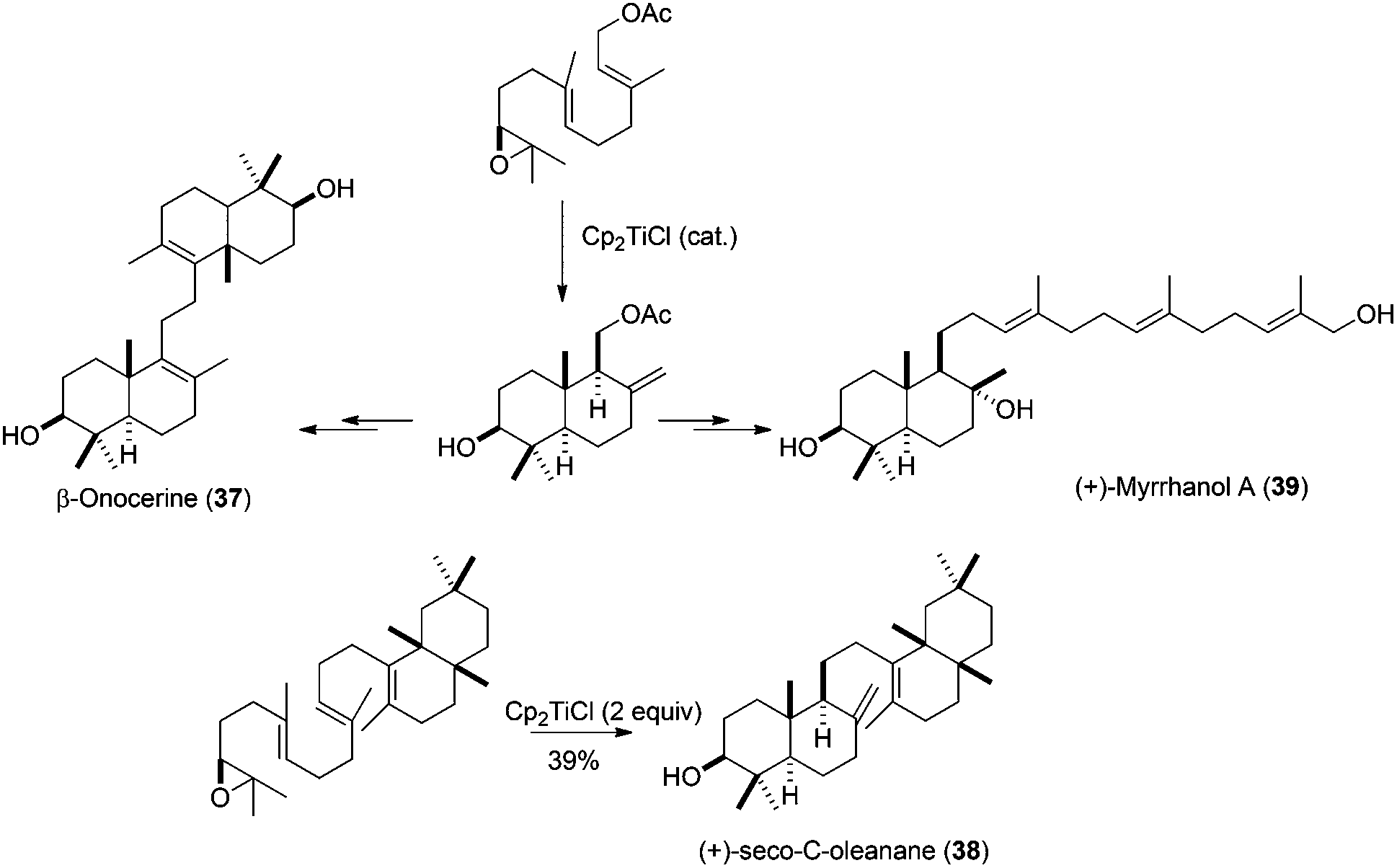 | ||
| Scheme 40 Synthesis of myrrhanol A, (+)-seco-C-oleanane, and β-onocerine via Cp2TiCl promoted epoxypolyene cyclisation. | ||
Sharpless epoxidation of allylic alcohols has been also used in the preparation of labdane-type compounds with complex structure.72 Another example of this application was the synthesis of tetracyclic fomitellic acid (40), a very interesting compound due to its biological activity as an inhibitor of calf DNA polymerase α, rat DNA polymerase β, and human DNA topoisomerases I and II.73 The synthesis of A,B rings of fomitellic acid was performed using a stereoselective Cp2TiCl-mediated cyclisation of the corresponding enantioenriched epoxide precursor (Scheme 41).
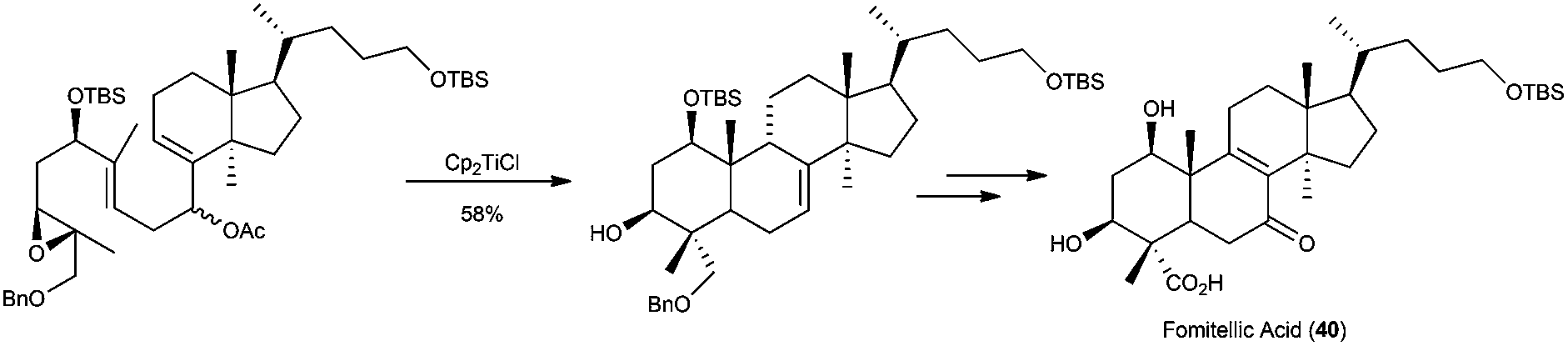 | ||
| Scheme 41 Approach to fomitellic acid based on a Cp2TiCl-mediated epoxypolyene cyclisation. | ||
Nevertheless, Sharpless dihydroxylation is not efficient in some substrates, presenting several problems concerning the enantiomeric purity as well as selectivity. To overcome these drawbacks, Cuerva et al. used Jacobsen epoxidation in the synthesis of the bicyclic natural terpene (−)-α-ambrinol (42),74 an odorous component of ambergris. The epoxidation of protected geranylacetone using this methodology, and subsequent cyclisation catalysed by Cp2TiCl, yielded the corresponding monocyclic product 41, which after a few simple steps was transformed in (−)-α-ambrinol (42) (Scheme 42).
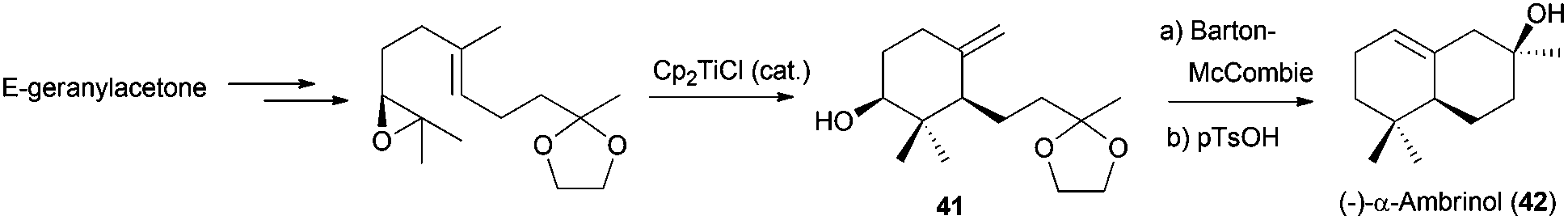 | ||
| Scheme 42 Synthetic approach to (−)-α-ambrinol. | ||
All the cyclisation processes depicted above are characterised by a 6-endo-trig cyclisation mode, similar to the related cationic processes. Nevertheless, in nature it is also possible to find compounds which present a cycloheptane in their structures, such as laukarlaool (44) or barekoxide (45). Additionally, tricyclic compounds from the valparane family also have this kind of seven-membered carbocycles. Although the biosynthesis of these compounds has been proposed to take place by sequence 6-endo, 6-endo, 7-endo cationic cyclisations, this biogenetic hypothesis involves the formation of secondary cationic intermediates.75 In fact, this kind of cationic cyclisation has not been reproduced in the laboratory even using enzymes,76 showing a lot of uncertainties about the real nature of 7-endo cyclisations. In this context, Cuerva's group has studied the cyclisation of several epoxypolyprenes derived from linalyl, nerolidyl and geranyl linalyl acyclic terpenes.77a The cyclisation of these epoxypolyprenes generated the corresponding cyclisation products presenting a 7-membered ring in good yields. These cyclisation products were subsequently used by the authors in the straightforward synthesis of several natural terpenes containing 7-membered rings, such as laukarlaool (44), barekoxide (45) and valparadiene (46). The syntheses were extremely short and effective. Thus for example, valparadiene (46) was prepared in only four steps from commercial geranyl linalool, with an overall yield of 21%. Recently, Cuerva et al. have studied the reason for this regioselectivity, concluding that the substitution in the double bond involved in the final cyclization reaction drives the reaction to the exclusive formation of six- or seven-membered rings (Schemes 43 and 44).77b
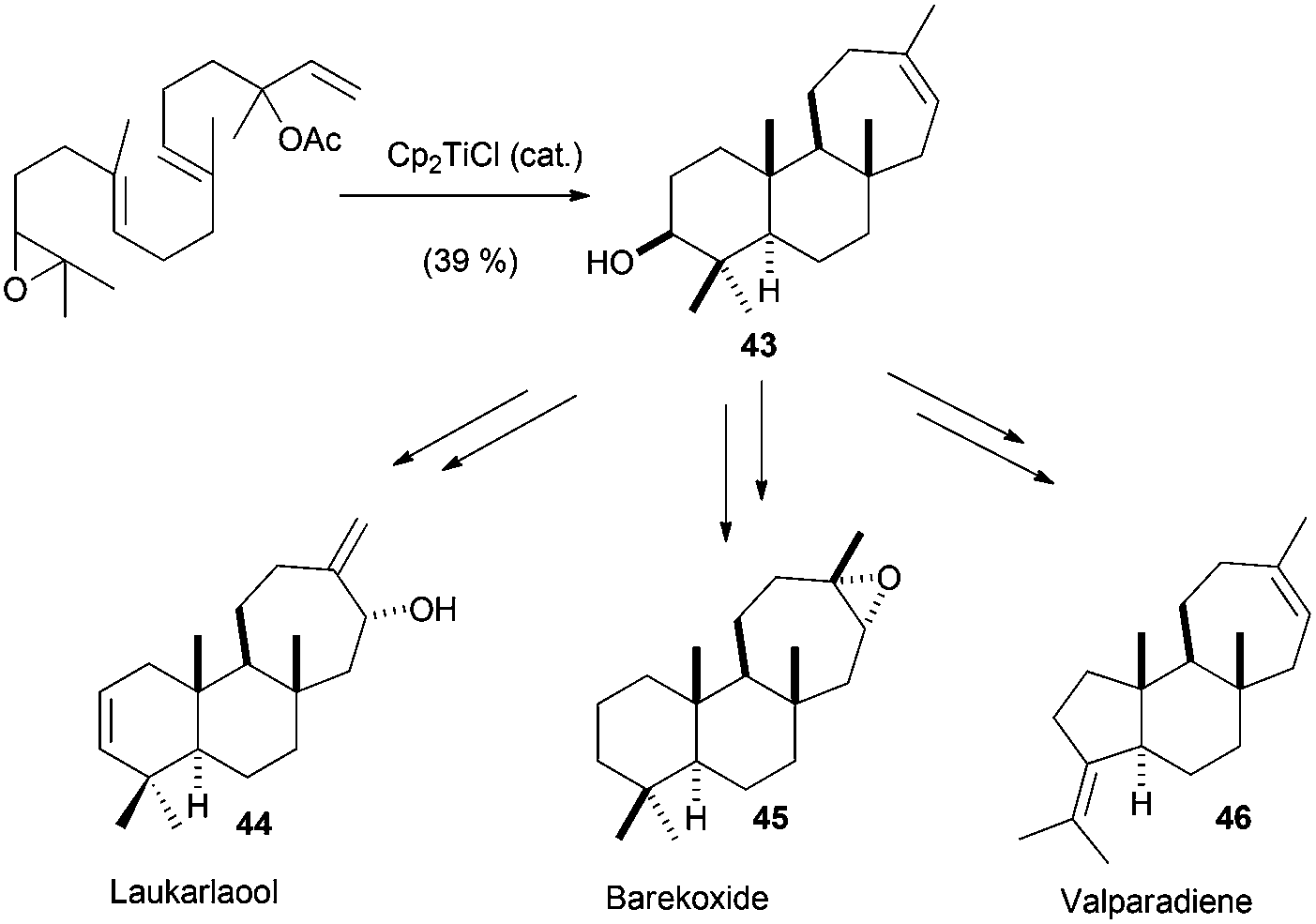 | ||
| Scheme 43 Cp2TiCl catalysed 7-endo cyclisations in epoxypolyene cyclisations. Synthesis of laukarlaool, barekoxide and valparadiene. | ||
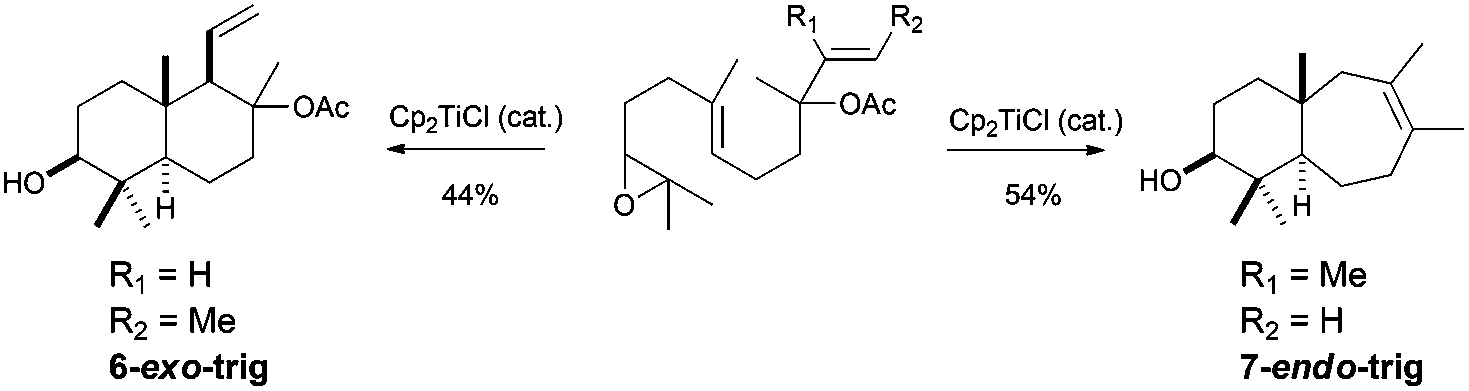 | ||
| Scheme 44 Selectivity in Cp2TiCl catalysed 6-exo vs. 7-endo cyclisations of epoxypolyenes. | ||
It is worth noting that the epoxide can be placed in other positions of the epoxypolyprene. Thus for example, Barrero et al. took advantage of this possibility in the preparation of chokols, a 2,6-cyclofarnesane compound isolated from Phleum pratense.78 The titanocene(III)-mediated cyclisation of an epoxide derivative of nerolidol led to the 5-exo-trig cyclisation product, used by the author in the synthesis of chokols derivatives (see Scheme 45). This unexpected cyclisation takes place due to a coordination between the titanocene complex, the epoxide and the trisubstituted hydroxyl group in vicinal position.
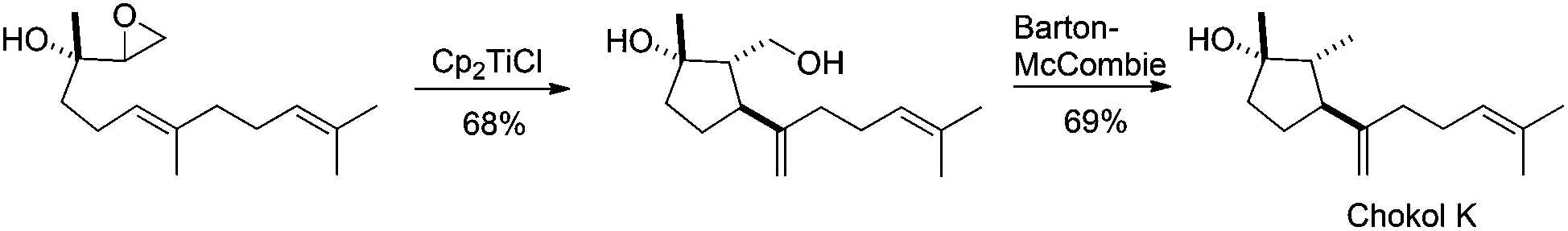 | ||
| Scheme 45 Synthesis of chokol K mediated by titanocene(III). | ||
The complex Cp2TiCl has not been only used in the synthesis of polycyclic terpenes with the regular skeletons shown before. It is also useful for the preparation of other terpenes with different structures. Thus, this methodology was used in the synthesis of eudesmanolide-type compounds, such as (+)-tuberiferine (50) and (+)-brachylaenolide (52), via titanocene(III)-catalysed cyclisation of the corresponding epoxygermacrolides.79a Additionally, an extensive study about the titanocene-promoted cyclization of structurally related epoxidated germacrones has been recently described (Scheme 46).79b
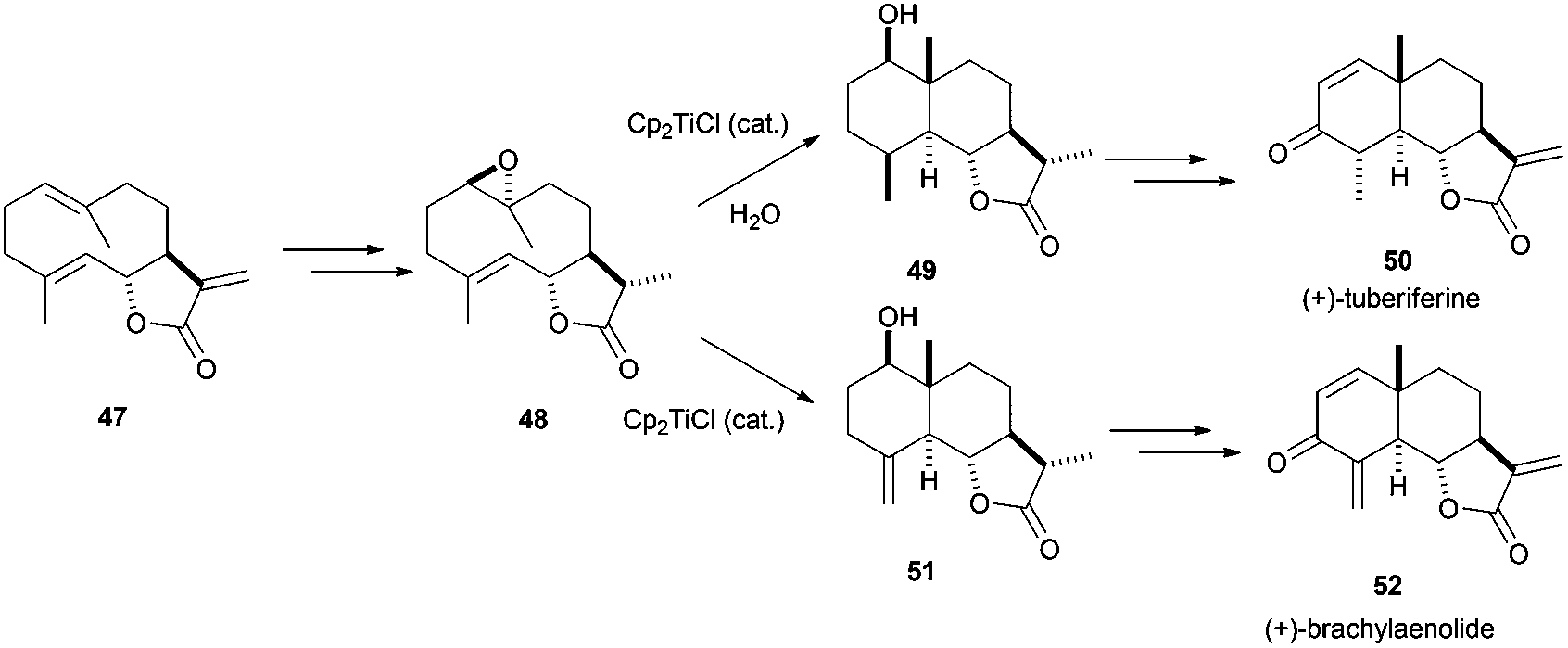 | ||
| Scheme 46 Preparation of (+)-tuberiferine (50) and (+)-brachylaenolide (52) by titanocene(III) chemistry. | ||
Recently, Fernández-Mateos et al. have performed a remarkable synthesis of CDE molecular fragments related to natural terpenoids sendanin, a limonoid triterpene,80a using stoichiometric amounts of Cp2TiCl (see Scheme 47). In this case, the epoxide is placed in a cyclic structure as 54, which is based on a terpene substructure derived from trimethylcyclohexenone 53. The subsequent cyclisation of 54 mediated by Cp2TiCl yields a polycyclic compound 55 with a structure similar to the CDE fragment present in the mentioned limonoid. A similar strategy has been used by the same authors in the synthesis of BCDE molecular fragments of the limonoid azadiradione.80b
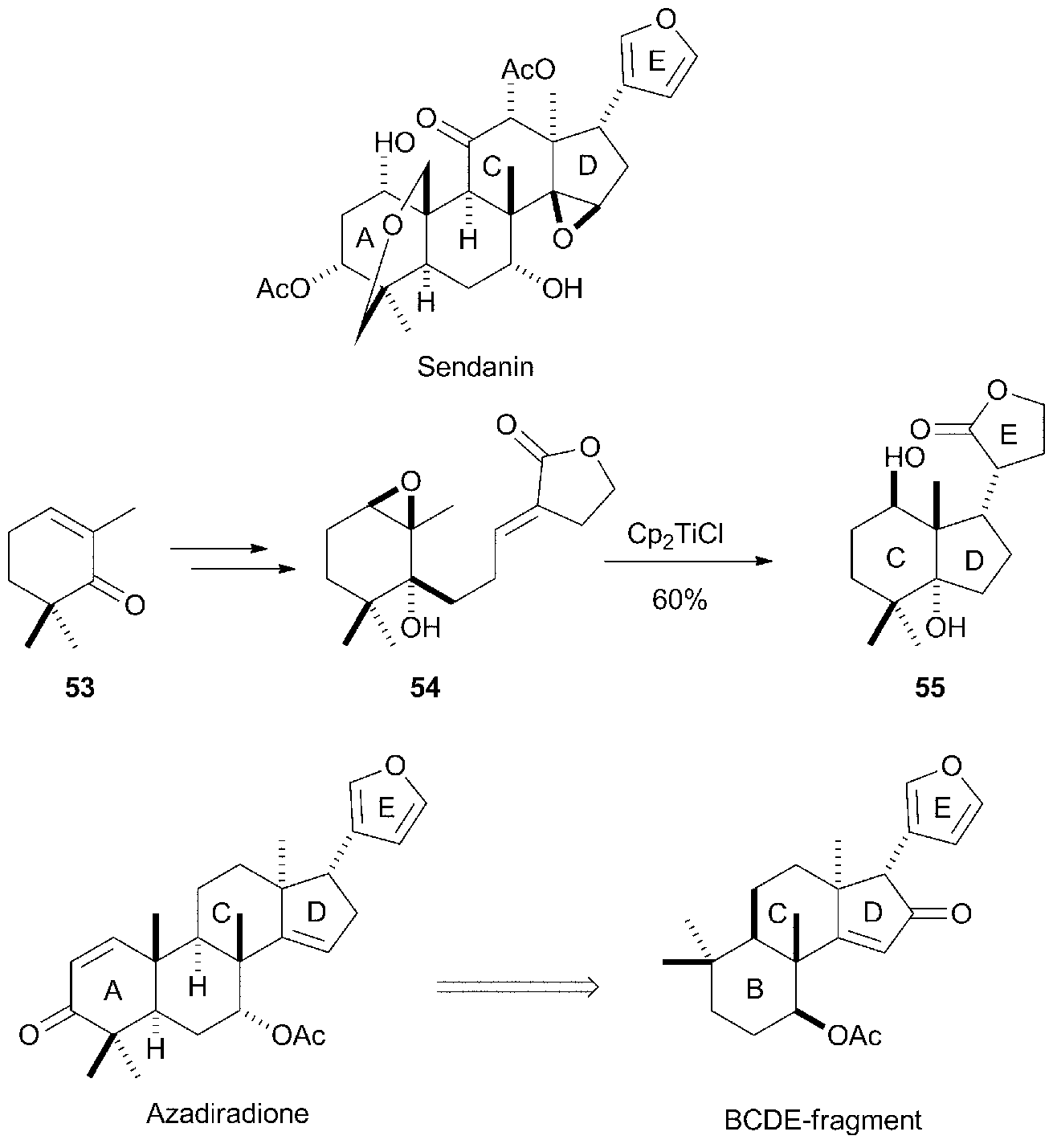 | ||
| Scheme 47 Synthesis of CDE fragment of sendanin, and BCDE fragment of azadiradione by Cp2TiCl-mediated cyclisation. | ||
Additionally, Gansäuer et al. described a new approach to the synthesis of fragranol, a monoterpene isolated from the roots of Artemisia fragrans Willd, based on a radical 4-exo-cyclisation catalysed by the new titanocene complex 56. This synthesis introduced a new concept in titanocene(III)-promoted cyclisations, a template effect from the complex to control the process (Scheme 48).81
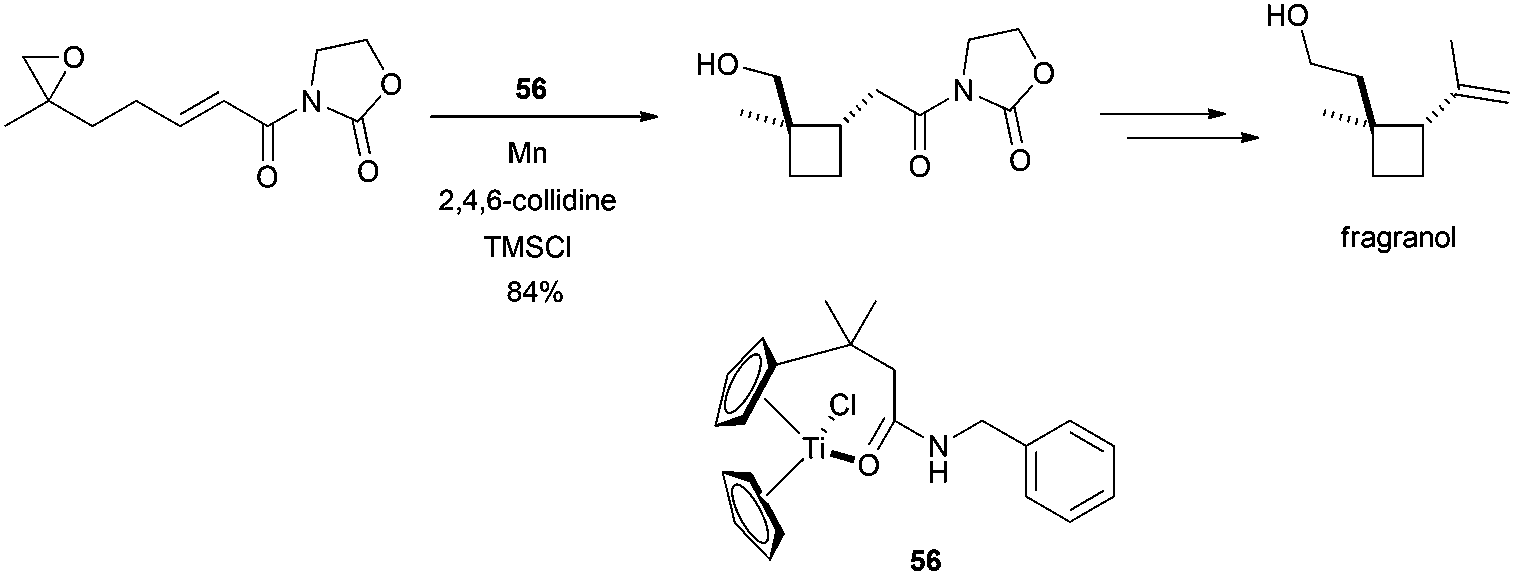 | ||
| Scheme 48 Synthesis of fragranol catalysed by a titanocene(III) complex 56. | ||
Other complex structures have been also prepared using titanocene(III)-promoted cyclisations. In this sense, Reisman's group has recently synthesised (−)-maoecrystal Z, an unusually rearranged 6,7-seco-ent-kauranoid natural terpenoid isolated from Isodon eriocalyx.82a In this synthesis, the authors used the ability of the radical generated from a Ti(III)-catalysed epoxide opening to give an addition reaction over acrylates, and the subsequent lactonisation reaction (Scheme 49). Additionally, an approximation to the synthesis of related compound maoecrystal V has recently been reported by Thomson's group.82b Other related compounds, such as (−)-trichorabdal A and (−)-longikaurin E, have been also prepared using this methodology.82c
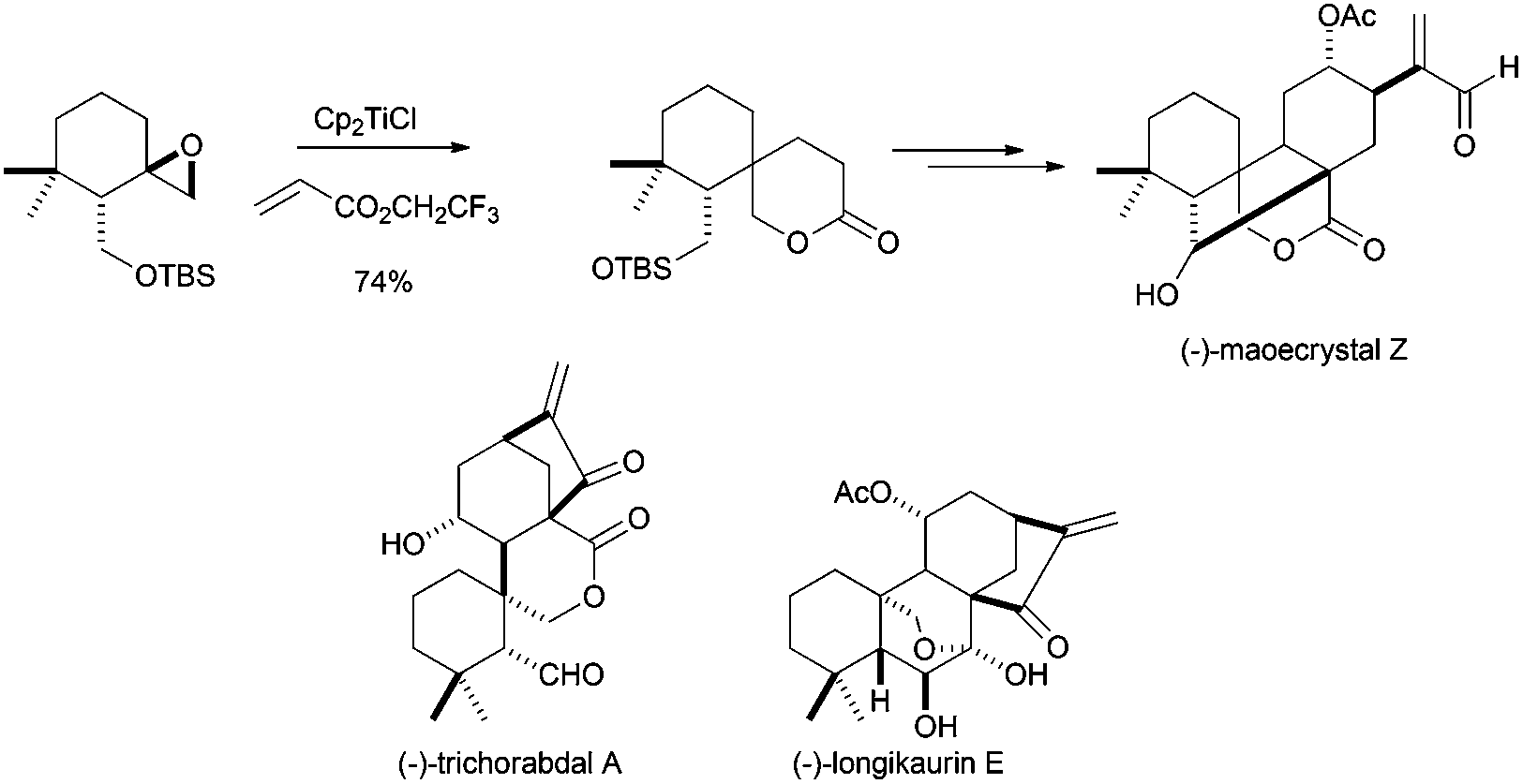 | ||
| Scheme 49 Synthesis of maoecrystal Z and related compounds via titanocene(III)-catalysed epoxide opening and lactonisation. | ||
Beyond the use of Cp2TiCl in the synthesis of cyclic or polycyclic terpenoids, acyclic terpenes can be also prepared. Thus for example, very efficient Wurtz- and Barbier-type reactions have been used in the synthesis of some natural acyclic terpenes. Cp2TiCl-catalyzed Wurtz-type coupling of farnesyl bromide has been used in the synthesis of squalene, an important natural terpene (Scheme 4).71 Recently, Cuerva et al. have published a modification of this process, using farnesyl carbonate as the starting material in a similar Wurtz-type coupling reaction catalysed by titanocene(III) and Pd(0).12a This new method allowed the use of more handle starting materials (Scheme 50).
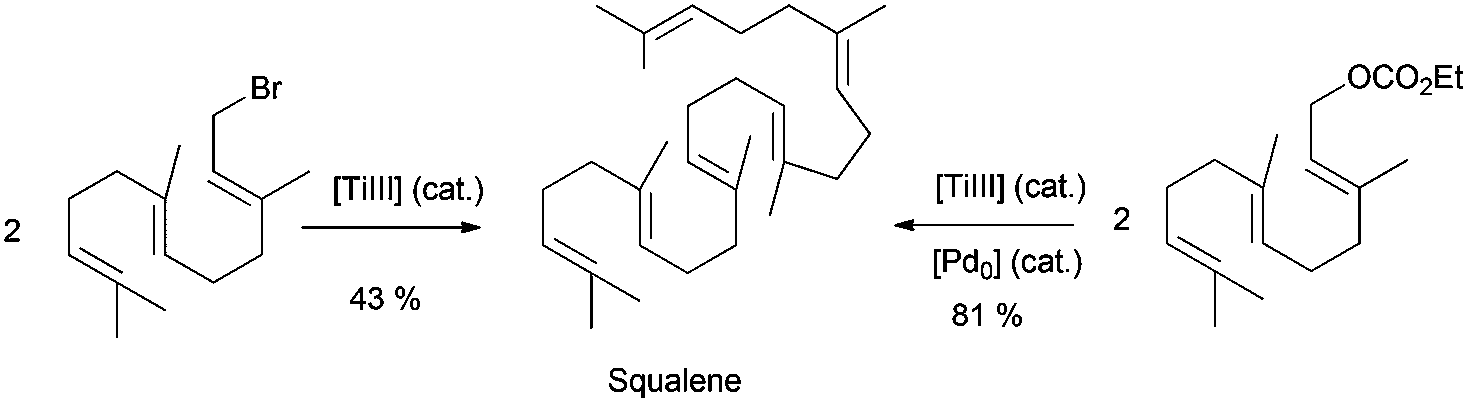 | ||
| Scheme 50 Synthesis of squalene by Wurtz type reactions from farnesyl bromide or farnesyl ethyl carbonate. | ||
Barbier-type reactions between prenyl halides and α,β-unsaturated aldehydes have also been used in the synthesis of hydroxylated acyclic terpenes (Scheme 4). In this sense, Cuerva's group performed the preparation of 12-hydroxysqualene, the major product from the biotransformation of presqualene diphosphate, and rosiridol, a monoterpenoid isolated from several plants, using these kinds of reactions (Scheme 51).6 Thus, 12-hydroxysqualene was synthesised in good yield by an α-regioselective Barbier reaction catalysed by titanocene(III) between farnesal and farnesyl chloride. On the other hand, rosiridol was prepared from prenyl bromide and the corresponding α,β-unsaturated aldehyde and subsequent transesterification.6
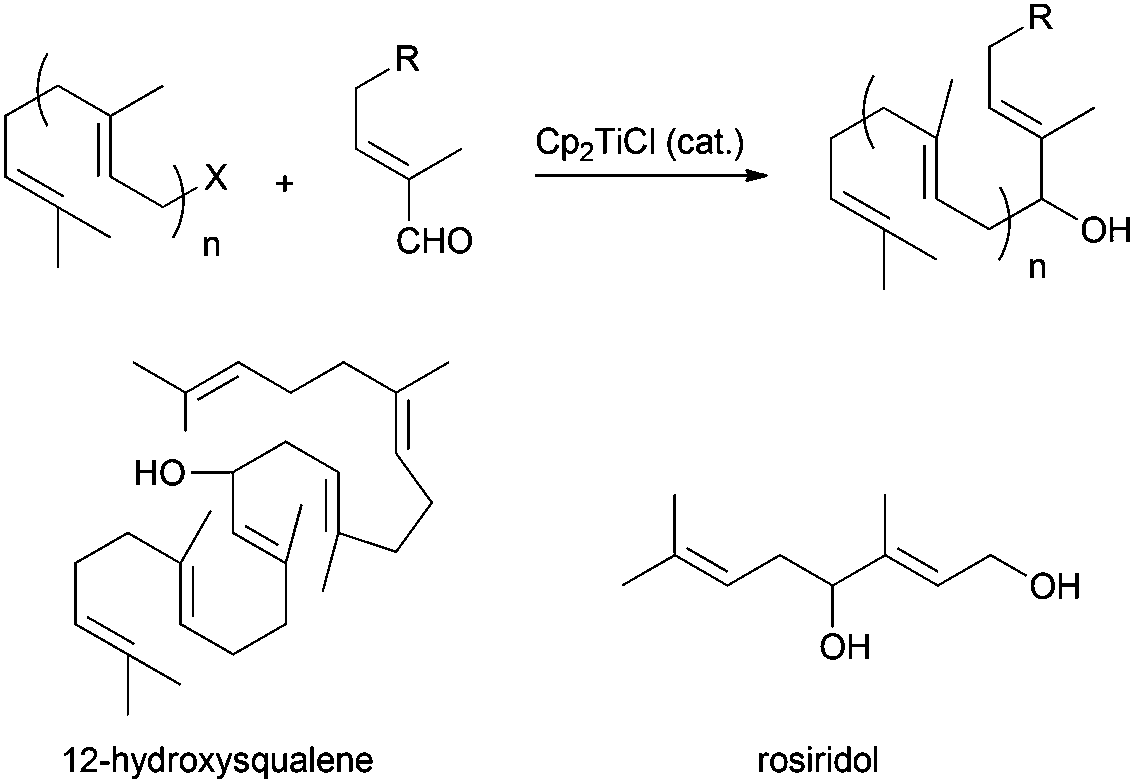 | ||
| Scheme 51 Preparation of acyclic hydroxylated terpenes using Cp2TiCl-catalyzed Barbier type reactions. | ||
All the chemistry shown above points out that Cp2TiCl is a powerful tool for the synthesis of natural acyclic and polycyclic terpenes, allowing access to this kind of compounds by straightforward strategies, using few synthetic steps and under mild conditions.
Meroterpenes
Meroterpenes are compounds of mixed biosynthesis with a common structural feature: they are formed by sesquiterpene units linked to aromatic structures, mostly substituted phenols. Most of the procedures described to synthesize these compounds using Cp2TiCl involve radical cyclizations starting from epoxipolyprenes and a great majority employ stoichiometric quantities of Ti(III).One example of this strategy was reported by Takahashi in the total synthesis of (±)-smenospondiol. The bicyclic skeleton was constructed by a Cp2TiCl-mediated 6-endo,6-exo tandem radical cyclization of an epoxyenyne, yielding the bicyclic structure as the major product (Scheme 52).83
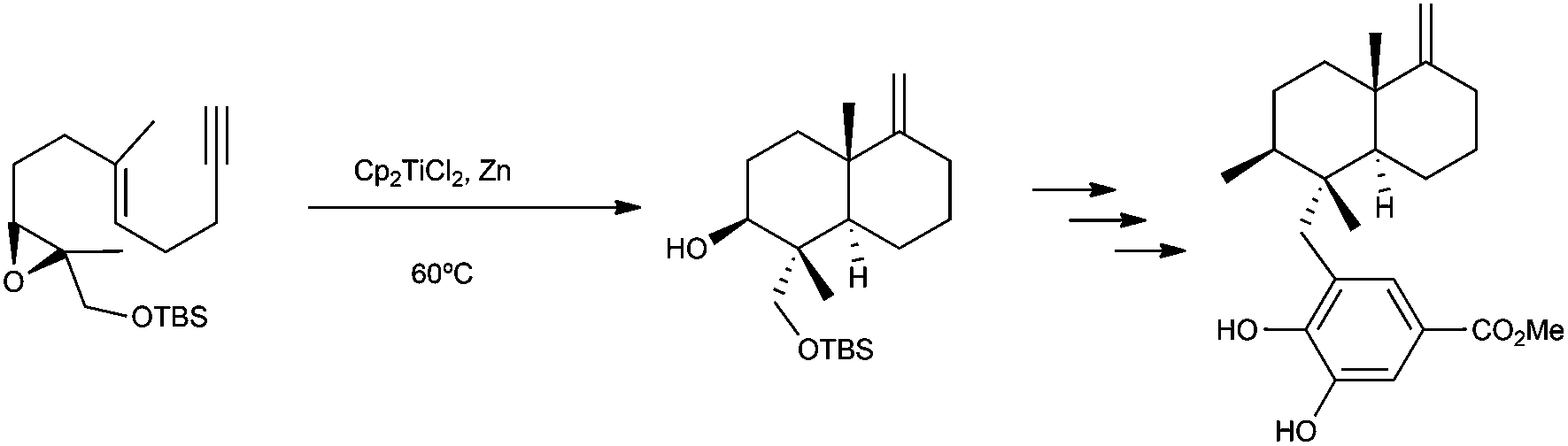 | ||
| Scheme 52 Synthesis of (±)-smenospondiol using titanocene(III) chemistry. | ||
Trost et al. used Cp2TiCl in the first enantioselective biomimetic total synthesis of (−)-sicannin. In this case, cationic cyclization using Lewis acids did not occur whereas Ti(III)-mediated cyclization afforded the desired tetracyclic compound 57 together with the 5-epi-sicannin in a 81% combined yield (Scheme 53).84
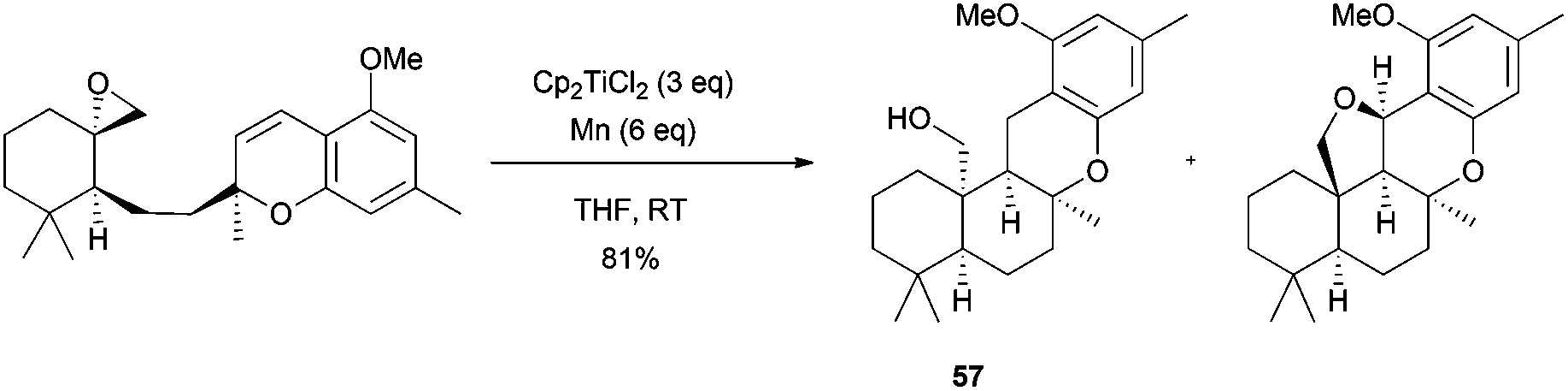 | ||
| Scheme 53 Preparation of 57, key intermediate in the synthesis of (−)-sicannin. | ||
The construction of the DEF-ring terpenoid fragment of terpendole E was achieved with a combination of Cp2TiCl2 and Zn, starting from an epoxi-derivative of the commercially available farnesyl acetate.85 In the total synthesis of (±)-platencin (58), Cp2TiCl promoted a highly stereoselective radical cyclization of a trisubstituted epoxide to an α,β-unsaturated ketone, yielding a single stereoisomer in 87% yield (Scheme 54).86
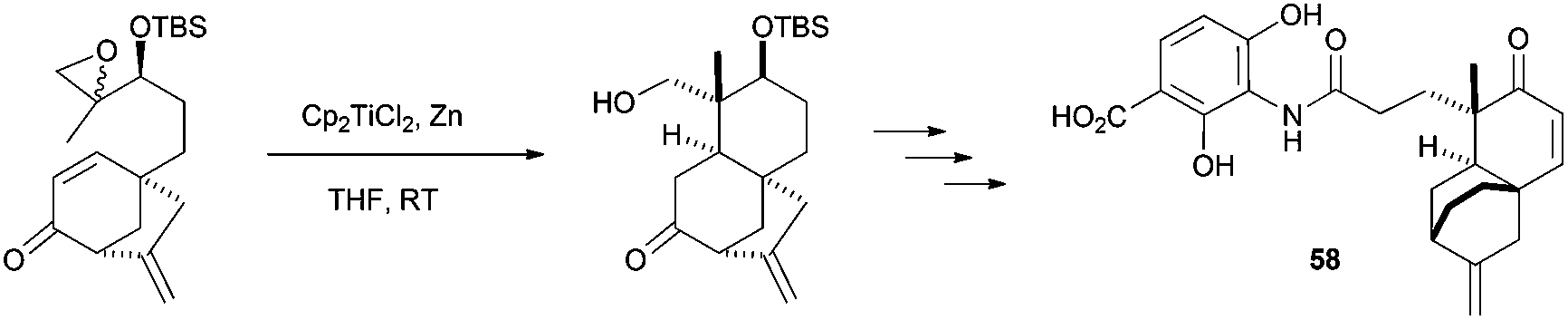 | ||
| Scheme 54 Synthesis of (±)-platencin (58) via Cp2TiCl-mediated cyclisation. | ||
The group of Omura and Nagamitsu proposed a stereoselective 6-exo-dig cyclization to a nitrile group, obtaining the corresponding diketoalcohol in 61% yield, which was later used in the total synthesis of pyripyropene A (Fig. 3).87
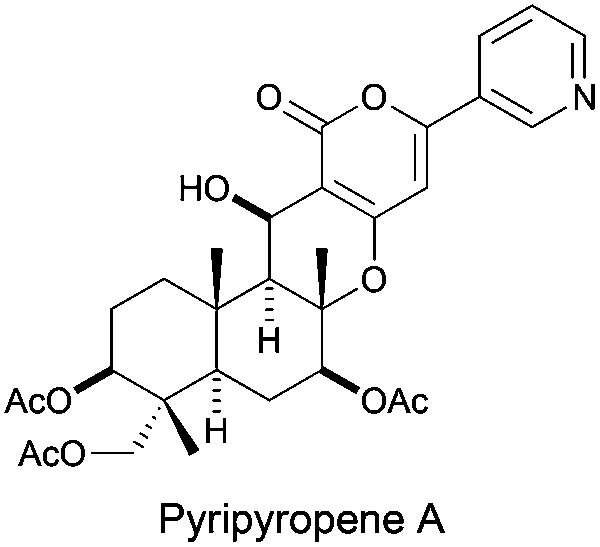 | ||
| Fig. 3 Structure of pyripyropene A. | ||
Recently, Yamashita et al. have described how Cp2TiCl is able to promote radical-induced atom transfer. In this case, the acryl ester moiety is transferred from an oxygen to a carbon, leading to the efficient construction of a quaternary center and the configurational inversion of C-9.88 Such a structure is an approximation to zoanthenol (59)(Scheme 55).
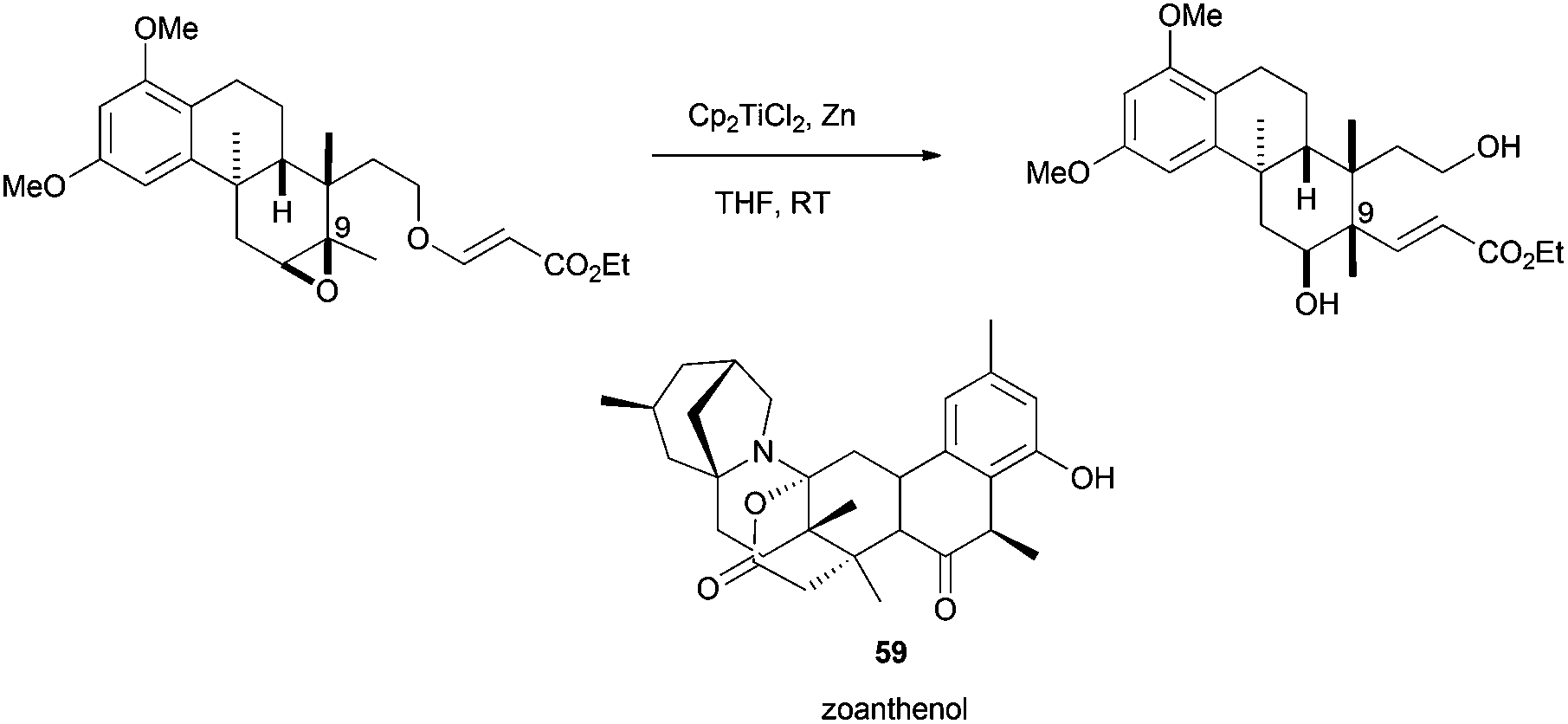 | ||
| Scheme 55 Approximation to the synthesis of zoanthenol (59). | ||
All the procedures described above involve the use of stoichiometric quantities of the titanium reagent. Nevertheless, the regenerating system introduced in our group, the chlorotrimethylsilane/2,4,6-collidine mixture, allowed the use of substoichiometric amounts of Ti(III). Thus, we have applied this methodology to the synthesis of different polycyclic meroterpenoids from aryl epoxypolyprenes (see Scheme 56), under mild conditions and with high stereo- and regioselectivities.89
 | ||
| Scheme 56 Straightforward access to meroterpenes. | ||
It is worth noting that the aromatic subunits remain unchanged after radical cyclization, which represents a significant difference not only to carbocationic processes but also to radical cyclizations promoted by other transition metals, such as Mn(III).
Antibiotics
Pharmaceuticals, and among them antibiotics, are another important group of compounds which have been synthesized using Cp2TiCl-mediated free radical chemistry. In this sense, the group of Roy has synthesised polysubstituted tetrahydrofuran anti-tumor antibiotics such as methylenolactocin (60),90 protolichesterinic acid (61),90 roccellaric acid (62),91 and dihydroprotolichesterinic acid (63)91 (Fig. 4).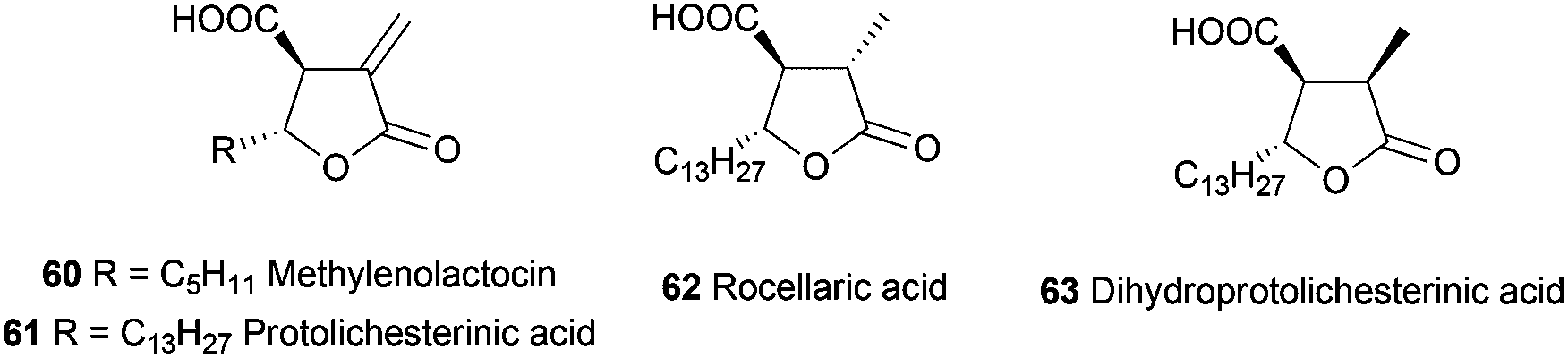 | ||
| Fig. 4 Anti-tumor antibiotics with tetrahydrofuran structures. | ||
The synthetic strategy for the preparation of 60 and 61 consists of an intramolecular radical cyclization of the epoxyalkyne derivatives 64 to give the tetrahydrofuran ring with a terminal alkene, which is characteristic of these antibiotics. (Scheme 57).
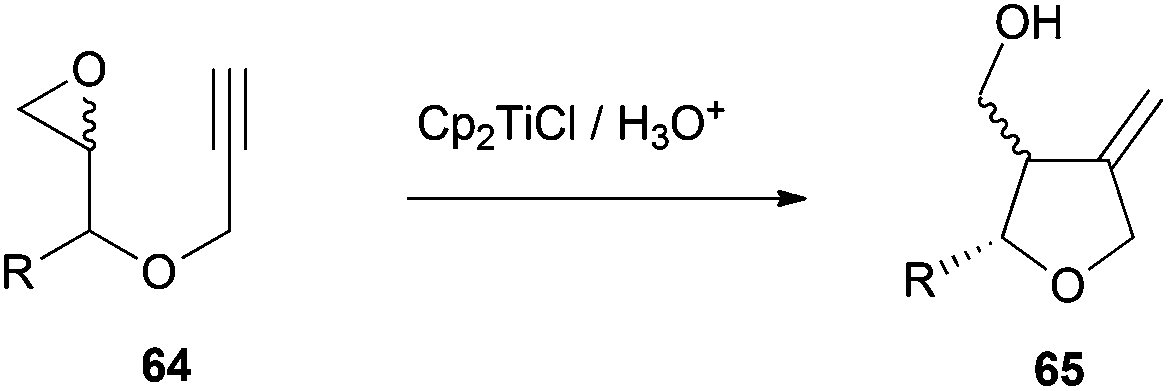 | ||
| Scheme 57 Preparation of intermediate 65via titanocene(III)-mediated cyclisation. | ||
The final compounds were obtained by a short sequence of steps, which implied protection of the hydroxyl group, oxidation with PDC to give the lactone, and deprotection and oxidation of the hydroxyl group to carboxylic acid with Jones reagent (Scheme 58).
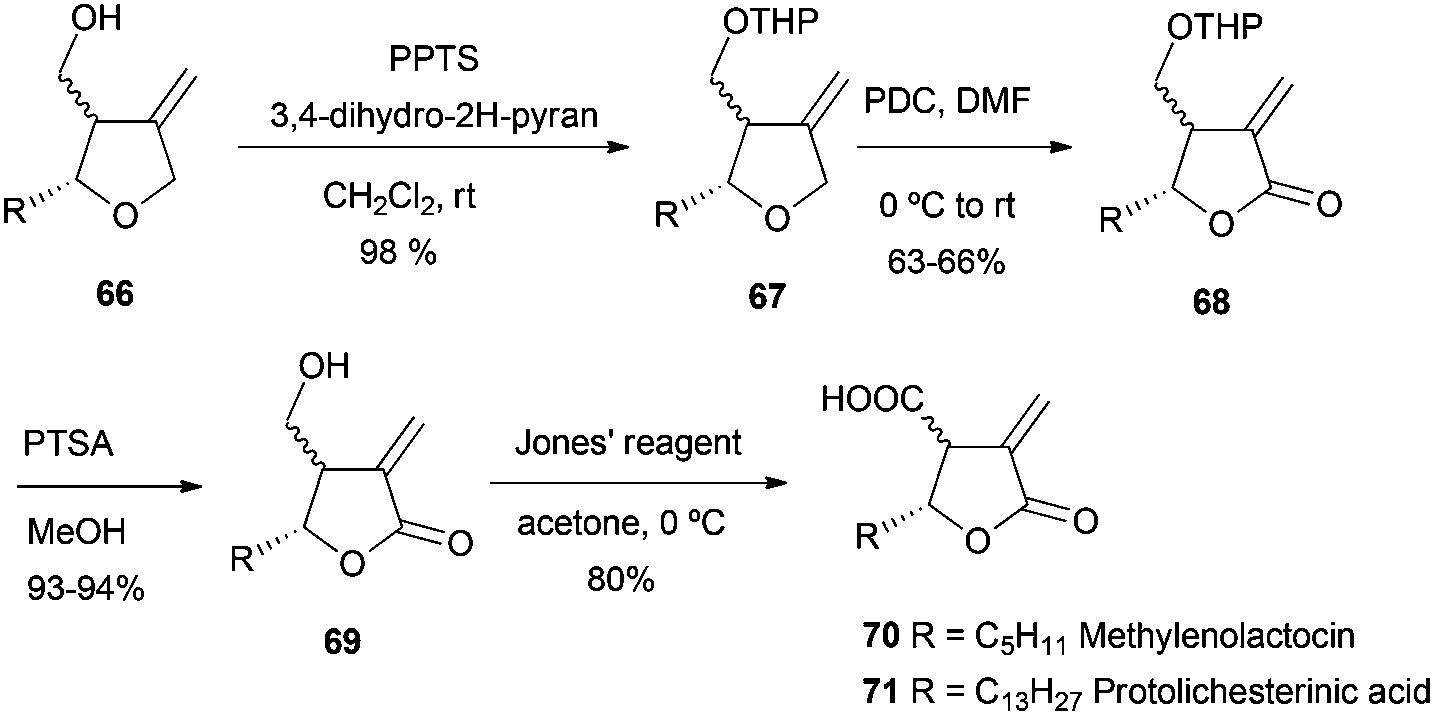 | ||
| Scheme 58 Synthesis of 70 and 71. | ||
The synthesis of 74 and 75 was carried out by an intramolecular radical cyclisation of the epoxyalkyne 72, followed by an oxidation with NaIO4 and a catalytic amount of RuCl3 to give the mixture of the final compounds (Scheme 59).
 | ||
| Scheme 59 Synthesis of 74 and 75. | ||
Recently, the same group has achieved the enantioselective synthesis of (−)-methylenolactocin and (−)-protolichesterinic acid starting with D-mannitol as a chiral precursor.92
Another group of antibiotics of broad spectrum and widely used are the β-lactam antibiotics, which contain a β-lactam ring in their molecular structures. The group of Grande has focused on the development of novel β-lactam derivatives by radical cyclisations using Cp2TiCl. A first approximation was the synthesis of highly functionalized chiral tricyclic β-lactams as very convenient intermediates to achieve chiral 3-substituted carbacephem antibiotics.93 The carbacephem skeletons were obtained by an intramolecular radical cyclisation of the corresponding enantiopure epoxides by addition to α,β-unsaturated lactones (Scheme 60).
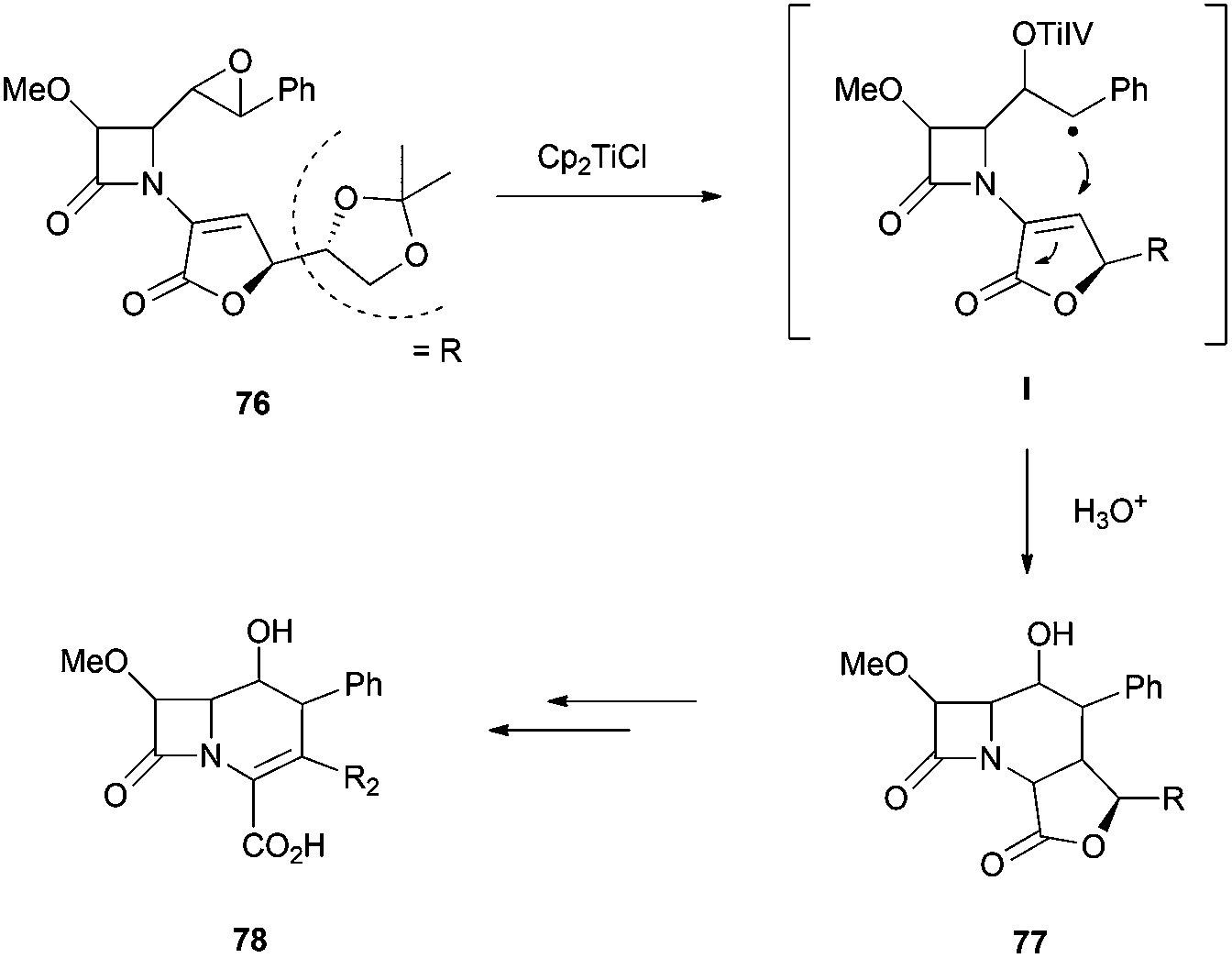 | ||
| Scheme 60 Synthesis of β-lactams skeletons mediated by Cp2TiCl. | ||
The reaction of the epoxy β-lactams with Cp2TiCl generated in situ with Cp2TiCl2 and zinc dust in THF afforded a mixture of three products in which the tricyclic β-lactams were the most abundant (Fig. 5).
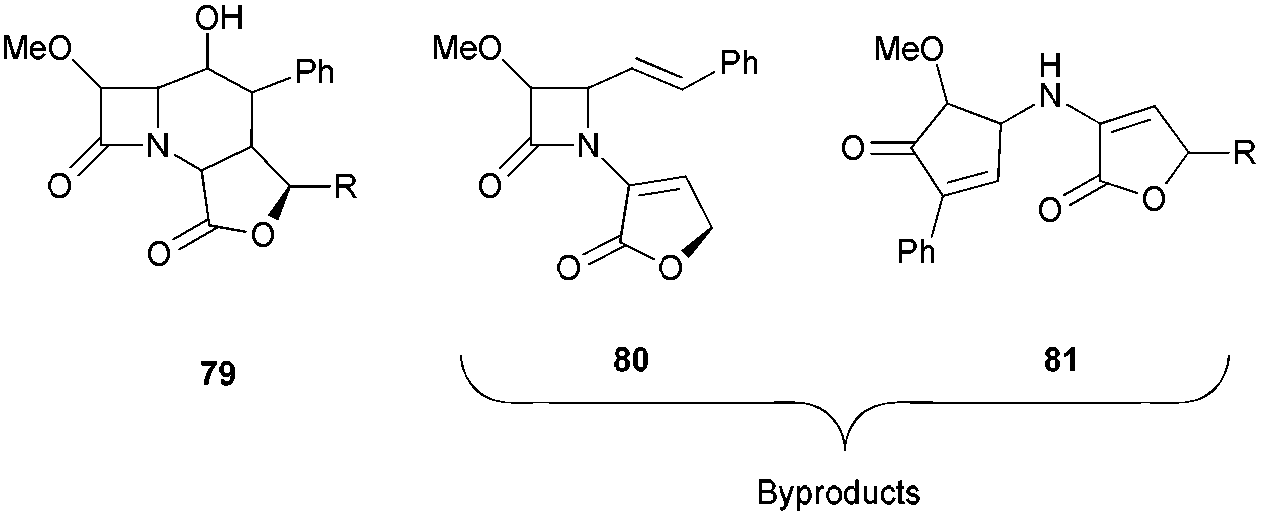 | ||
| Fig. 5 Mixture of products in the cyclisation step. | ||
This strategy has been extended to the synthesis of stereospecific polyfunctionalized bicyclic β-lactams. In this case, the intramolecular radical cyclisation mediated by Cp2TiCl was conducted over four isomeric epoxyacrylates 82 and epoxyaldehydes 83. While the four isomeric epoxyaldehydes gave exclusively the respective carbacephems 85, under the same reaction conditions, only two isomeric epoxyacrylates gave the cyclisation products 84ab and 84ba together with the elimination products (all isomers)94 (Scheme 61).
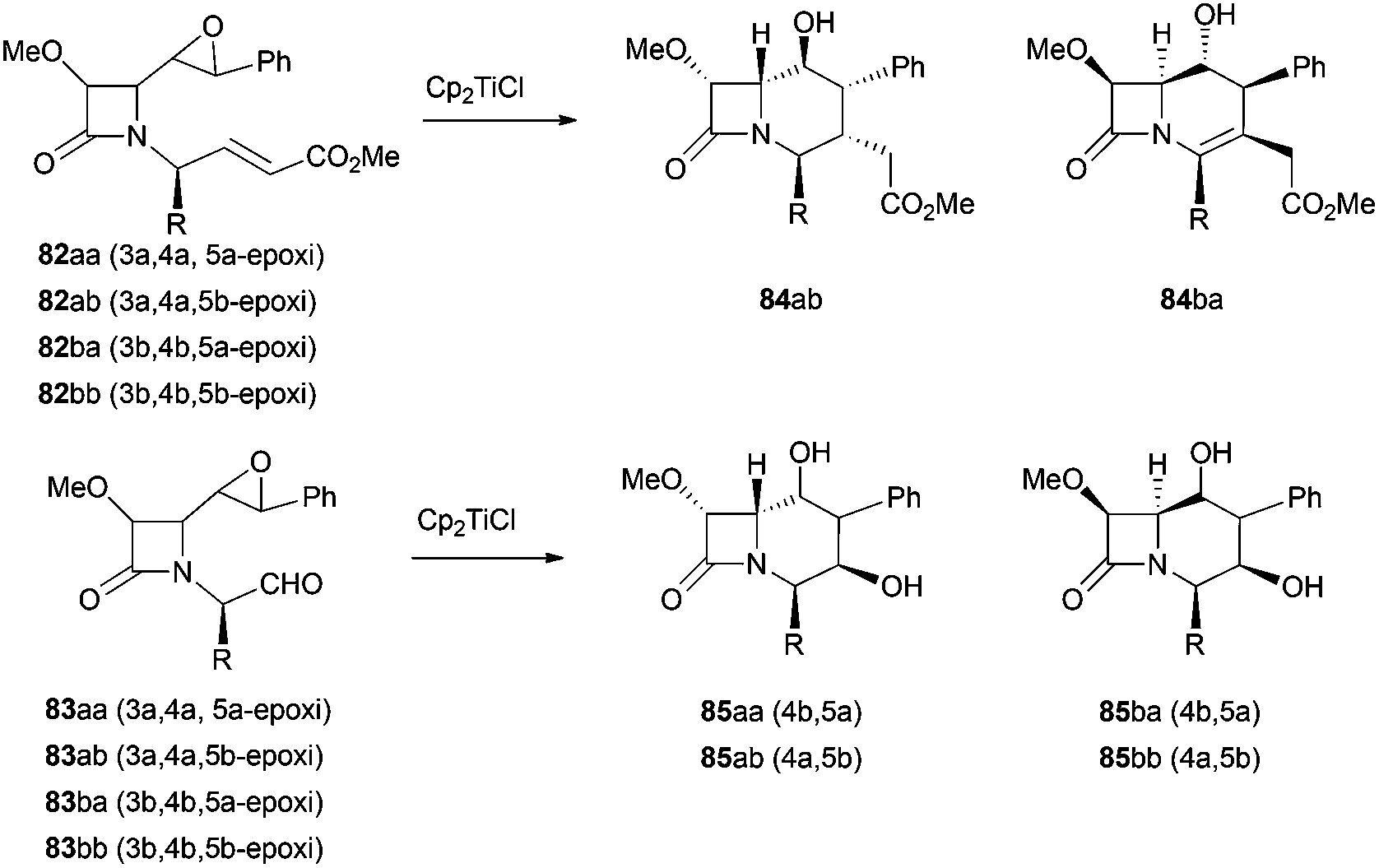 | ||
| Scheme 61 Synthesis of novel carbacephems. | ||
More recently, the same group has reported the synthesis of new bi- and tricyclic β-lactams by the radical cyclisation of δ- and ε-epoxynitrile-2-azetidinones 86 and 87. The intramolecular radical cyclisation between the homolytic opening of the epoxide and the cyano group afforded, after hydrolysis, bi- and tricyclic hydroxyketo-β-lactams95 (Scheme 62).
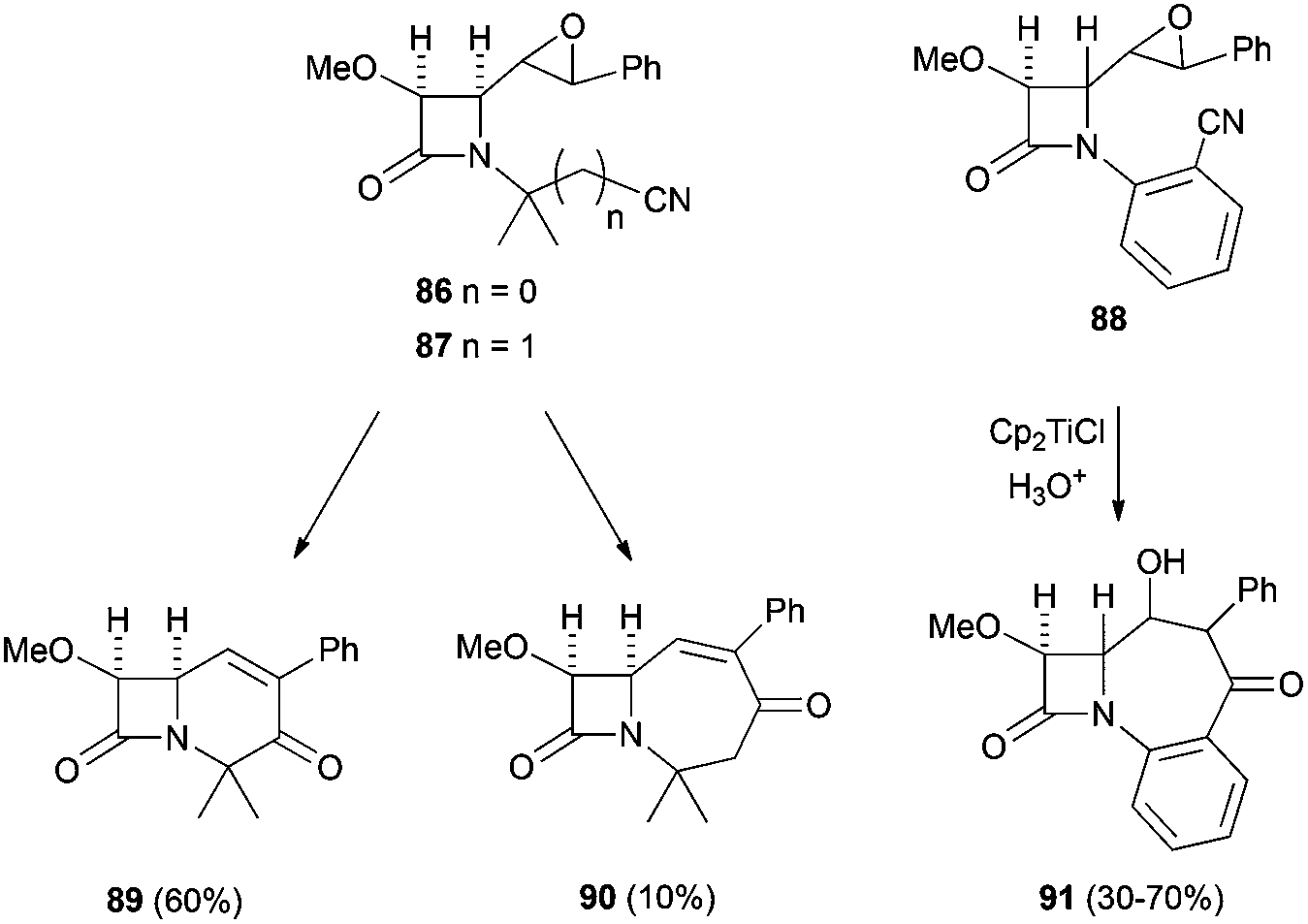 | ||
| Scheme 62 Cp2TiCl mediated radical cyclisation of δ- and ε-epoxynitrile-2-azetidinones. | ||
The authors proved that a 7-exo-radical cyclisation of ε-epoxynitrile-2-azetidinones promoted by Cp2TiCl is possible and new tricyclic β-lactams could be synthesised. This reaction was applied for the first time to benzonitrile acceptors to give the first examples of homobenzocarbacephems 91 reported in the literature.95a
Miscellaneous
Another group of natural products which have been synthesized using Cp2TiCl as a radical initiator are 3,4-dihydroisocumarins. The group of Roy has achieved the synthesis of hydrangenol 97, phyllodulcin 98, macrophyllol 99, and thunberginol G 100.96 The mixture of the ethyl 2-bromomethyl-benzoate derivative 92 and the corresponding benzaldehyde (93–96) was treated with Cp2TiCl in THF at room temperature to afford the corresponding 3-phenyl-3,4-dihydroisocoumarins in good yield. The reaction proceeded via a Barbier-type addition followed by in situ lactonization (Scheme 63).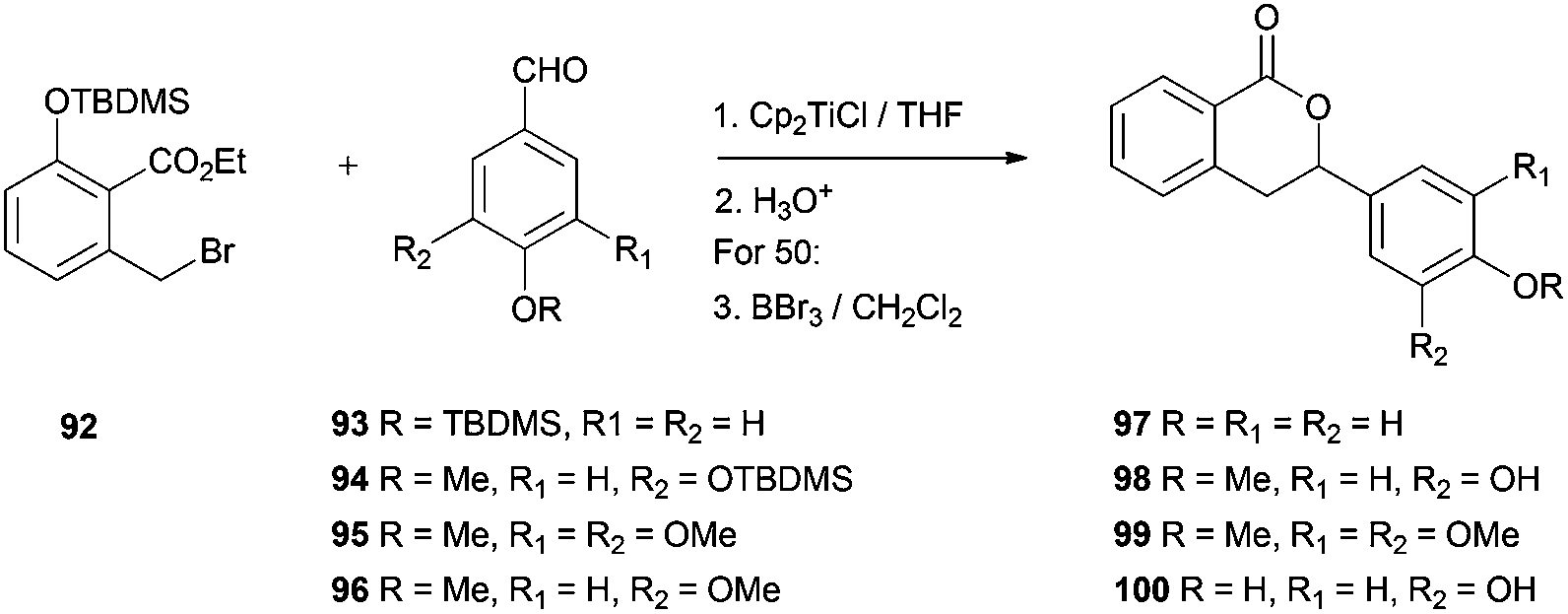 | ||
| Scheme 63 Synthesis of 3,4-dihydroisocumarins. | ||
Additionally to these interesting substances, Cp2TiCl has also been used for the synthesis of polyfunctionalized carbo- and heterocyclic precursors of many natural products. For example, polyfunctionalized tetrahydrofurans and pyrans97 have been synthesized by radical cyclisation reactions of epoxyallenes98 and epoxyacrylates ethers (Scheme 64).97
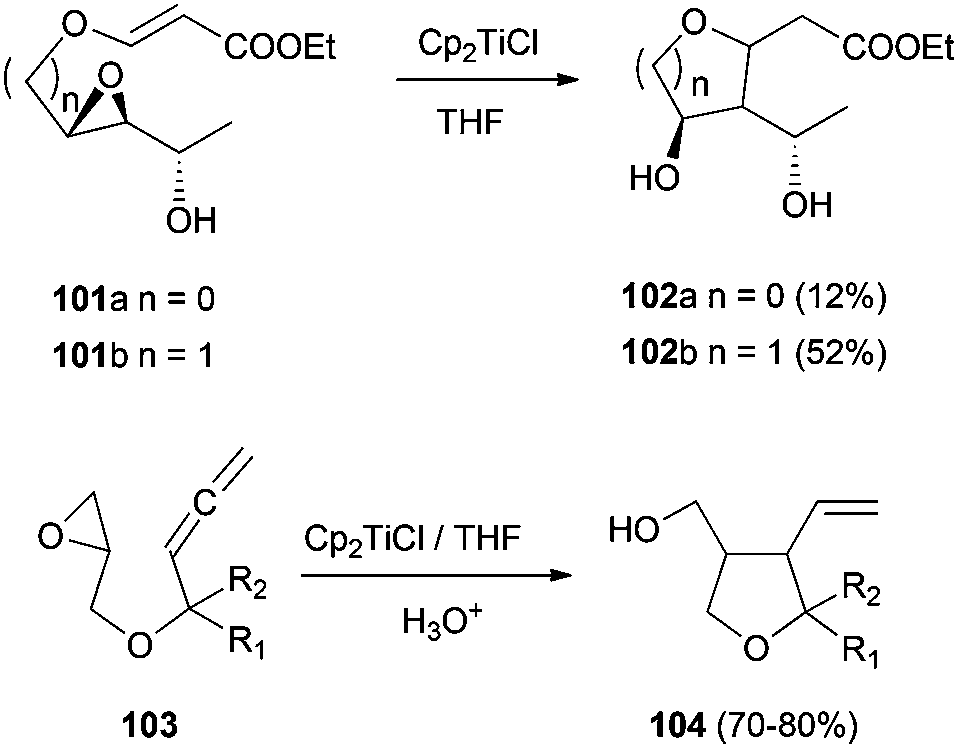 | ||
| Scheme 64 Synthesis of polyfunctionalised tetrahydrofurans. | ||
Five-membered carbocycles with multiple chiral centres have been achieved by intramolecular radical cyclisation of the corresponding enantiopure epoxyaldehydes99 and epoxyacrylates (Scheme 65).100 This strategy has been used in the total synthesis of 2-epirosmarinecine (Scheme 66).101
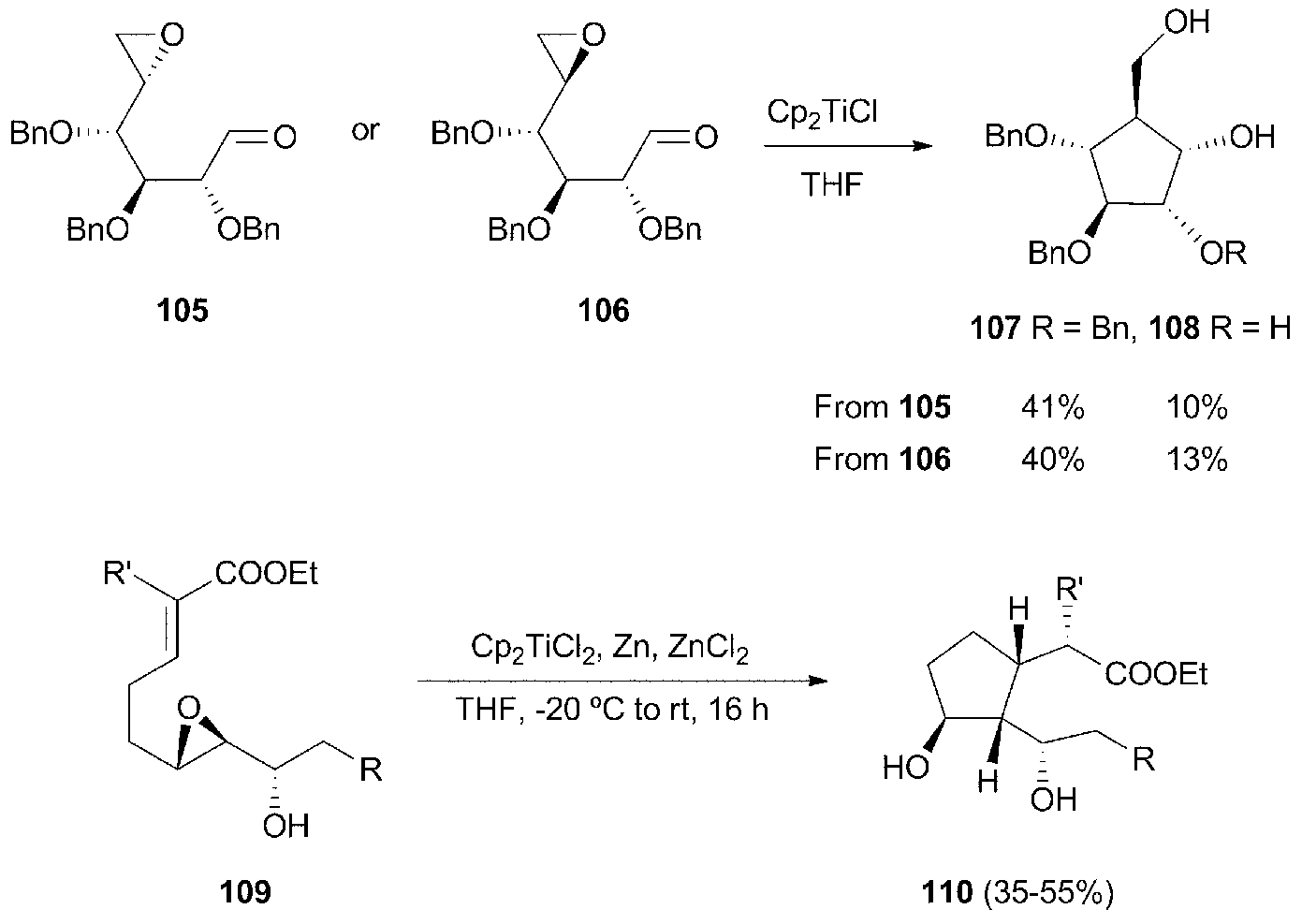 | ||
| Scheme 65 Synthesis of chiral cyclopentanols. | ||
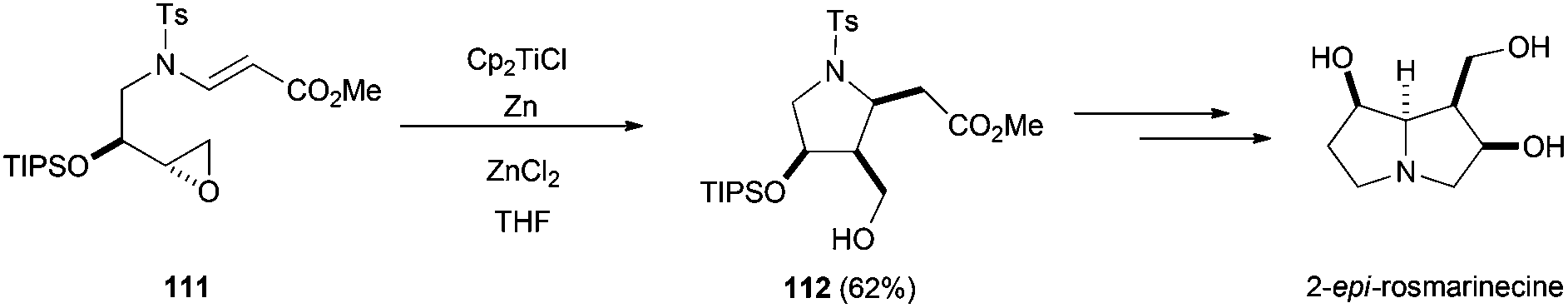 | ||
| Scheme 66 Synthesis of 2-epirosmarinecine. | ||
Another class of important building blocks present in a multitude of natural products are oxacyclic fused rings, such as bicyclic ethers and bis-γ-lactones. The synthesis of both types of structures has been carried out by the group of Roy. The synthesis of the bicyclic ethers 113 proceeded via intramolecular radical cyclisation of the corresponding epoxyalkynes 112.102 The synthesis of the bis-γ-lactones 116 was achieved by an intramolecular Barbier type addition of 114 followed by PDC oxidation103 (Scheme 67).
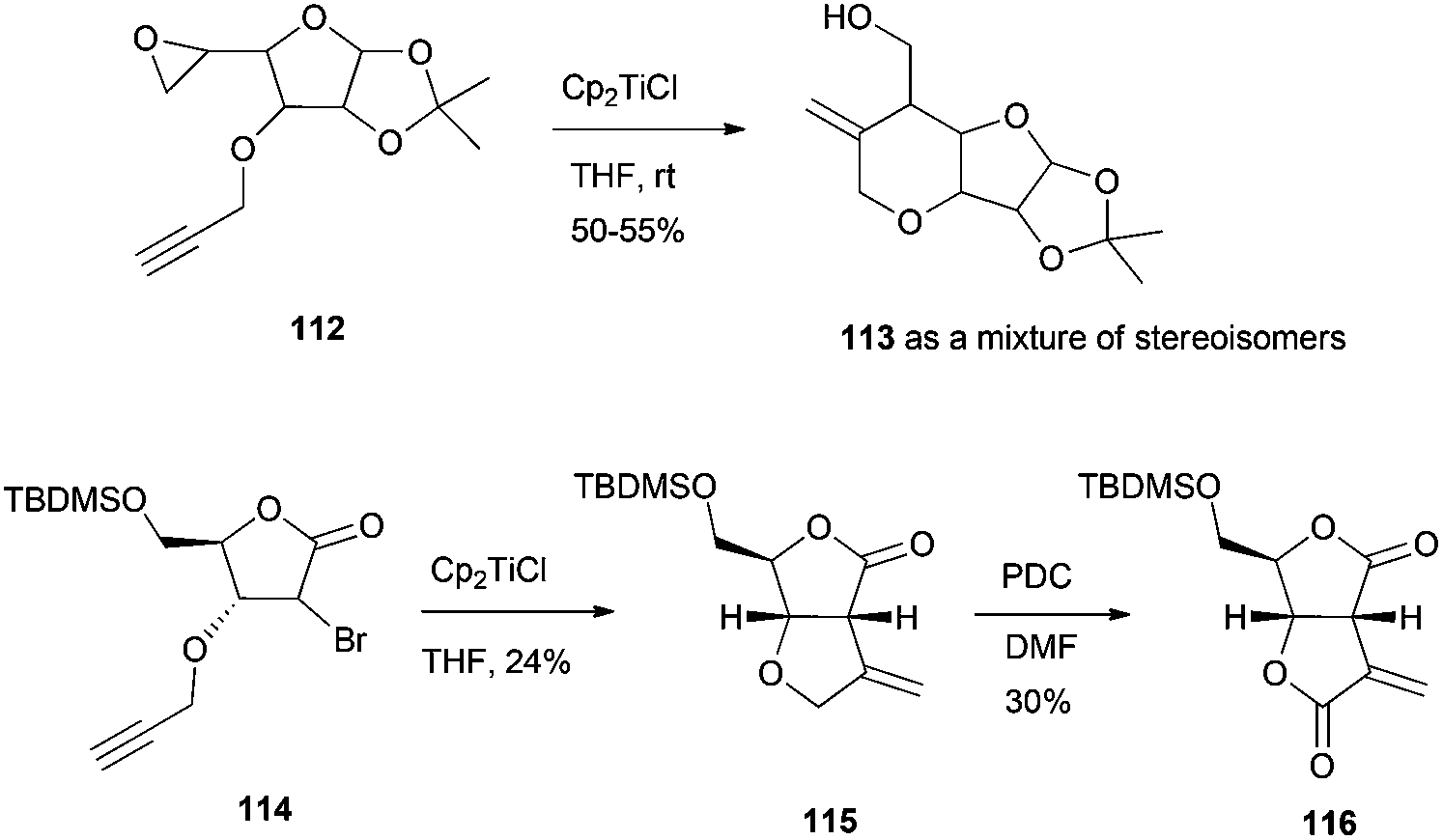 | ||
| Scheme 67 Synthesis of oxacyclic fused rings. | ||
Roy et al. synthesized substituted furans from α-bromo-β-keto enolethers using Cp2TiCl as a radical source. This methodology was applied to the synthesis of evodone, a furan monoterpene (Scheme 68).104
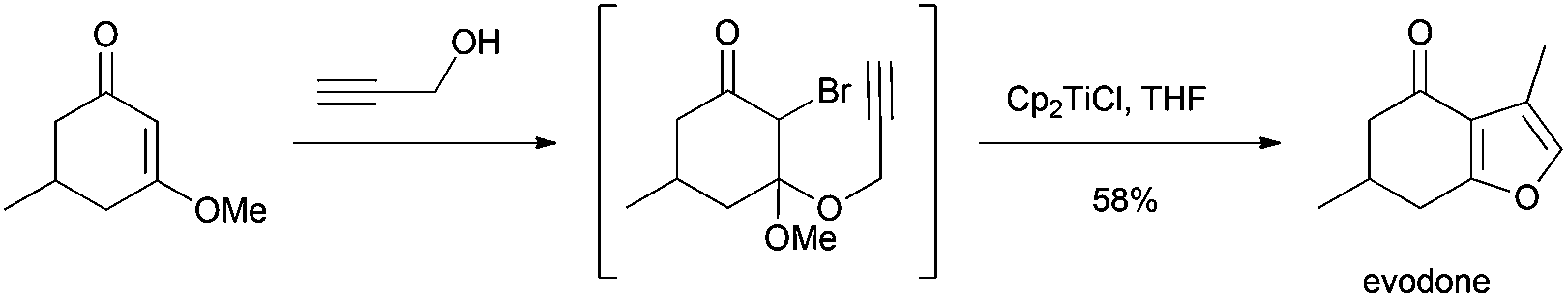 | ||
| Scheme 68 Synthesis of evodone. | ||
Recently, the synthesis of (±)-9,10-diepi-stemoamide 118 has been reported by the group of Zheng. The key step for the synthesis of this compound is a novel dehydroxylative radical cyclisation with the activated alkene 117 catalysed by Cp2TiCl105 (Scheme 69).
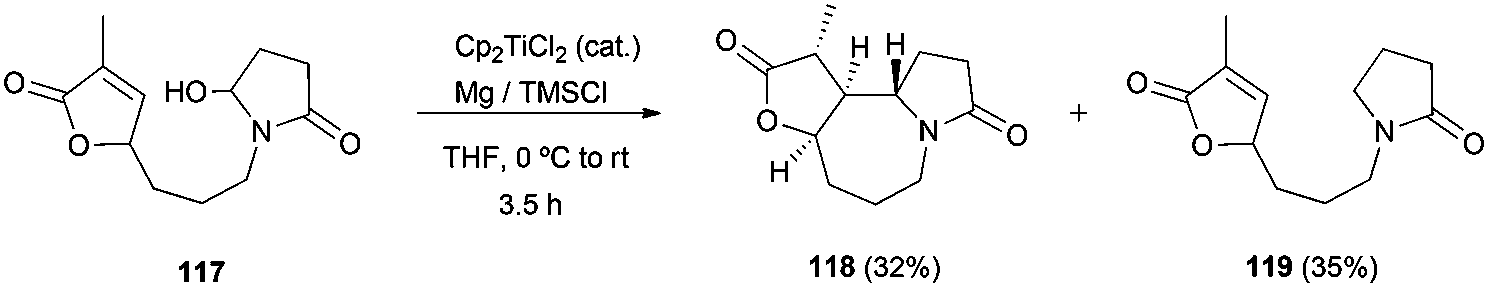 | ||
| Scheme 69 Synthesis of (±)-9,10-diepi-stemoamide 118. | ||
Other important kinds of bioactive natural compounds, such as α-aminated ketones (as 122) and pyrrolidin-3-ones, are also accessible using titanocene(III) catalytic cyclization of imines over nitrile moieties, as has been recently demonstrated by Streuff et al. (Scheme 70).106 This methodology is also useful in the preparation of α-hydroxyketone fragments, present in several natural compounds as barbacenic acid, cortistatin D, dragmacidin F, sieboldine A, etc.107
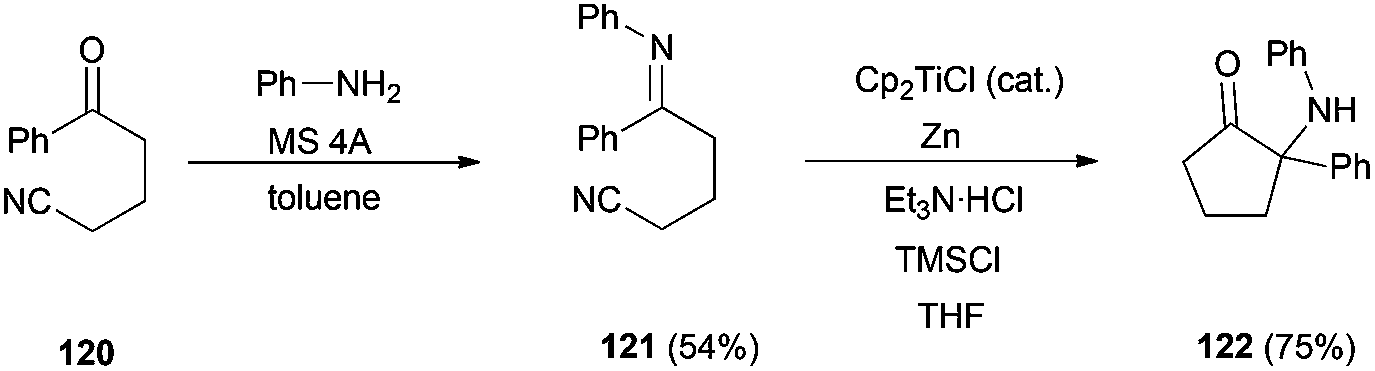 | ||
| Scheme 70 Synthesis of α-aminated ketone 122. | ||
Cp2TiCl-catalysed intramolecular radical addition of an epoxide to an alkyne has been used in the recent synthesis of the important drug entecavir, frequently used in the treatment of hepatitis B. The introduction of IrCl(CO)(PPh3)2/H2 in the reaction conditions allowed the reduction of the final radical (Scheme 71).108
 | ||
| Scheme 71 Ti(III)-catalysed synthesis of 124, an intermediate in the preparation of entecavir. | ||
Conclusions
Titanocene(III)-mediated radical processes have been applied to the synthesis of natural products of diverse nature. Beyond simple functional group interconversions, radical cyclizations, mainly from epoxides, have demonstrated their utility to yield (poly)cyclic natural skeletons, which are valuable synthons in organic synthesis. This radical approach has in many cases resulted in better yields and stereoselectivities than the cationic equivalents. In particular, the synthesis at room temperature of stereodefined terpenic skeletons without enzymatic assistance is remarkable. In this context, the main limitation of this bioinspired approach is, in fact, its extraordinary stereoselectivity, which avoids obtaining cis-fused decalins and/or substituents in axial positions. Such stereochemistry is present in many interesting natural terpenes. On the other hand, as can be seen in the first part of the review, the diverse reactivity of titanocene(III) complexes derives in some functional group incompatibilities. It is expected that in near future judicious designs of new titanocene(III) complexes can resolve this drawback. In any case, the evolution of the applications of titanocene(III) in natural product synthesis suggests that these reagents can be a matter of choice in the arsenal of a synthetic organic chemist.Acknowledgements
We thank the Regional Government of Andalucía (project P09-FQM-4571), MICINN (project CTQ-2011.22455), and CEI-Biotic for financial support. SPM thanks the Regional Government of Andalucía for her FPI fellowship. DM and AGC thank the Regional Government of Andalucía and the MICINN (Juan de la Cierva) for their postdoctoral contracts.Notes and references
- T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986 CrossRef CAS.
- A. Gansäuer, M. Pierobon and H. Bluhm, J. Am. Chem. Soc., 1998, 120, 12849 CrossRef.
- (a) A. Hafner, R. O. Duthaler, R. Marti, G. Rihs, P. Rothe-Streit and F. Schwarzenbach, J. Am. Chem. Soc., 1992, 114, 2321 CrossRef CAS; (b) A. Bensari, J. L. Renaud and O. Riant, Org. Lett., 2001, 3, 3863 CrossRef CAS PubMed; (c) Z. Chen and R. L. Halterman, J. Am. Chem. Soc., 1992, 114, 2276 CrossRef CAS; (d) E. Cesarotti, H. B. Kagan, R. Goddard and C. Krüger, J. Organomet. Chem., 1978, 162, 297 CrossRef CAS; (e) F. R. W. P. Wild, L. Zsolnai, G. Huttner and H. H. Brintzinger, J. Organomet. Chem., 1982, 232, 233 CAS.
- (a) A. Gansäuer, J. Justicia, C.-A. Fan, D. Worgull and F. Piestert, Top. Curr. Chem., 2007, 279, 25 CrossRef; (b) A. F. Barrero, J. F. Quílez del Moral, E. M. Sánchez and J. F. Arteaga, Eur. J. Org. Chem., 2006, 1627–1641 CrossRef CAS; (c) J. M. Cuerva, J. Justicia, J. L. Oller-López and J. E. Oltra, Top. Organomet. Chem., 2006, 63–91 CAS; (d) B. Rossi, S. Prosperini, N. Pastori, A. Clerici and C. Punta, Molecules, 2012, 17, 14700–14732 CrossRef CAS PubMed; (e) A. Gansäuer and A. Fleckhaus, Epoxides in titanocenes-mediated and –catalyzed radical reactions, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, UK, 2012, vol. 2, pp. 989–1001 Search PubMed.
- A. Gansäuer, N. Ndene, T. Lauterbach, J. Justicia, I. Winkler, C. Mück-Lichtenfeld and S. Grimme, Tetrahedron, 2008, 64, 11839 CrossRef PubMed.
- R. E. Estévez, J. Justicia, B. Bazdi, N. Fuentes, M. Paradas, D. Choquesillo-Lazarte, J. M. García-Ruiz, R. Robles, A. Gansäuer, J. M. Cuerva and J. E. Oltra, Chem.–Eur. J., 2009, 15, 2774 CrossRef PubMed.
- A. F. Barrero, M. M. Herrador, J. F. Quílez del Moral, P. Arteaga, J. F. Arteaga, M. Piedra and E. M. Sánchez, Org. Lett., 2005, 7, 2301 CrossRef CAS PubMed.
- (a) J. D. Parrish, D. R. Shelton and R. D. Little, Org. Lett., 2003, 5, 3615 CrossRef CAS PubMed; (b) R. E. Estévez, M. Paradas, A. Millán, T. Jiménez, R. Robles, J. M. Cuerva and J. E. Oltra, J. Org. Chem., 2008, 73, 1616 CrossRef PubMed.
- (a) J. M. Cuerva, A. G. Campaña, J. Justicia, A. Rosales, J. L. Oller–López, R. Robles, D. J. Cárdenas, E. Buñuel and J. E. Oltra, Angew. Chem., Int. Ed., 2006, 45, 5522 CrossRef CAS PubMed; (b) M. Paradas, A. G. Campaña, M. L. Marcos, J. Justicia, A. Haidour, R. Robles, D. J. Cárdenas, J. E. Oltra and J. M. Cuerva, Dalton Trans., 2010, 39, 8796 RSC; (c) H. R. Diéguez, A. López, V. Domingo, J. F. Arteaga, J. A. Dobado, M. M. Herrador, J. F. Quílez del Moral and A. F. Barrero, J. Am. Chem. Soc., 2010, 132, 254 CrossRef PubMed.
- (a) A. Gansäuer, Chem. Commun., 1997, 457 RSC; (b) A. Gansäuer, M. Moschioni and D. Bauer, Eur. J. Org. Chem., 1998, 1923 CrossRef; (c) A. Gansäuer and D. Bauer, Eur. J. Org. Chem., 1998, 2673 CrossRef; (d) A. Gansäuer and D. Bauer, J. Org. Chem., 1998, 63, 2070 CrossRef; (e) M. Paradas, A. G. Campaña, R. E. Estévez, L. Álvarez de Cienfuegos, T. Jiménez, R. Robles, J. M. Cuerva and J. E. Oltra, J. Org. Chem., 2009, 74, 3616 CrossRef CAS PubMed; (f) J. Streuff, Synthesis, 2013, 281–307 CrossRef CAS PubMed.
- J. Justicia, A. Rosales, E. Buñuel, J. L. Oller-López, M. Valdivia, A. Haïdour, J. E. Oltra, A. F. Barrero, D. J. Cárdenas and J. M. Cuerva, Chem.–Eur. J., 2004, 10, 1778 CrossRef CAS PubMed.
- (a) A. G. Campaña, B. Bazdi, N. Fuentes, R. Robles, J. M. Cuerva, J. E. Oltra, S. Porcel and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 7515 CrossRef PubMed; (b) A. Millán, A. G. Campaña, B. Bazdi, D. Miguel, L. Álvarez de Cienfuegos, A. M. Echavarren and J. M. Cuerva, Chem.–Eur. J., 2011, 17, 3985 CrossRef PubMed; (c) A. Millán, L. Álvarez de Cienfuegos, A. Martín-Lasanta, A. G. Campaña and J. M. Cuerva, Adv. Synth. Catal., 2011, 353, 73 CrossRef; (d) A. Millán, A. Martín-Lasanta, D. Miguel, L. Álvarez de Cienfuegos and J. M. Cuerva, Chem. Commun., 2011, 47, 10470 RSC; (e) A. Millán, L. Álvarez de Cienfuegos, D. Miguel, A. G. Campaña and J. M. Cuerva, Org. Lett., 2012, 14, 5984 CrossRef PubMed.
- J. Justicia, T. Jiménez, S. P. Morcillo, J. M. Cuerva and J. E. Oltra, Tetrahedron, 2009, 65, 10837 CrossRef CAS PubMed.
- T. V. Rajanbabu, W. A. Nugent and M. S. Beattie, J. Am. Chem. Soc., 1990, 112, 6408 CrossRef CAS.
- T. Golakoti, J. Ogino, C. E. Heltzel, T. L. Husebo, C. M. Jensen, L. K. Larsen, G. M. L. Patterson, R. E. Moore, S. L. Mooberry, T. H. Corbett and F. A. Valeriote, J. Am. Chem. Soc., 1995, 117, 12030 CrossRef CAS.
- (a) C. Hardouin, E. Doris, B. Rousseau and C. Mioskowski, Org. Lett., 2002, 4, 1151–1153 CrossRef CAS PubMed; (b) C. Hardouin, E. Doris, B. Rousseau and C. Mioskowski, J. Org. Chem., 2002, 67, 6571 CrossRef CAS PubMed.
- (a) C. L. Cavallaro and J. Schwartz, J. Org. Chem., 1995, 60, 7055 CrossRef CAS; (b) R. P. Spencer and J. Schwartz, Tetrahedron, 2000, 56, 2103 CrossRef CAS.
- T. Hansen, K. Daasbjerg and T. Skrydstrup, Tetrahedron Lett., 2000, 41, 8645 CrossRef CAS.
- J. S. Yadav, T. Shekharam and V. R. Gadgil, J. Chem. Soc., Chem. Commun., 1990, 843 RSC.
- A. V. R. Rao, M. N. Bhanu and G. V. M. Sharma, Tetrahedron Lett., 1993, 34, 707 CrossRef CAS.
- Z. Liu and R. Bittman, Org. Lett., 2012, 14, 620 CrossRef CAS PubMed.
- J. S. Yavav, D. Srinivas and T. Shekharam, Tetrahedron Lett., 1994, 35, 3625 CrossRef.
- (a) F. Bermejo and C. Sandoval, J. Org. Chem., 2004, 69, 5275 CrossRef CAS PubMed; (b) A. Fernández-Mateos, P. Herrero Teijón and R. Rubio González, Tetrahedron, 2013, 69, 1611 CrossRef PubMed.
- A. Fernández-Mateos, P. Herrero and R. Rubio, Tetrahedron, 2011, 67, 9259 CrossRef PubMed.
- K. V. Bhaskar and L. M. Mander, Tetrahedron Lett., 1996, 37, 719 CrossRef CAS.
- Q. Yanlong, L. Guisheng and Y.-Z. Huang, J. Organomet. Chem., 1990, 381, 29 CrossRef.
- K. B. Jørgensen, T. Suenaga and T. Nakata, Tetrahedron Lett., 1999, 40, 8855 CrossRef.
- T. K. Chakraborty and S. Tapadar, Tetrahedron Lett., 2001, 42, 1375 CrossRef CAS.
- T. K. Chakraborty, A. K. Chattopadhyay and S. Ghosh, Tetrahedron Lett., 2007, 48, 1139 CrossRef CAS PubMed.
- J. S. Yadav, T. Shekharam and D. Srinivas, Tetrahedron Lett., 1992, 33, 7973 CrossRef CAS.
- C. Hardouin, F. Chevallier, B. Rousseau and E. Doris, J. Org. Chem., 2001, 66, 1046 CrossRef CAS.
- L. Moisan, C. Hardouin, B. Rousseau and E. Doris, Tetrahedron Lett., 2002, 43, 2013 CrossRef CAS.
- A. D. Kosal and B. L. Ashfeld, Org. Lett., 2010, 12, 44 CrossRef CAS PubMed.
- A. Fernández-Mateos, P. Herrero-Teijón, R. Rabanedo-Clemente and R. Rubio-González, Tetrahedron Lett., 2009, 47, 7755 CrossRef PubMed.
- T. K. Chakraborty and S. Das, Tetrahedron Lett., 2002, 43, 2313 CrossRef CAS.
- T. K. Chakraborty, R. Samanta and S. Das, J. Org. Chem., 2006, 71, 3321 CrossRef CAS PubMed.
- J. D. Parrish and D. R. Little, Org. Lett., 2002, 4, 1439 CrossRef CAS PubMed.
- G. A. Nishiguchi and R. D. Little, J. Org. Chem., 2005, 70, 5249 CrossRef CAS PubMed.
- T. Jiménez, A. G. Campaña, B. Bazdi, M. Paradas, D. Arráez-Román, A. Segura-Carretero, A. Fernández-Gutiérrez, J. E. Oltra, R. Robles, J. Justicia and J. M. Cuerva, Eur. J. Org. Chem., 2010, 4288 CrossRef.
- S. Diethelm, C. S. Chindler and E. M. Carreira, Org. Lett., 2010, 12, 3950 CrossRef CAS PubMed.
- S. Jana, C. Guin and S. C. Roy, J. Org. Chem., 2005, 70, 8252 CrossRef CAS PubMed.
- S. Saha and S. C. Roy, J. Org. Chem., 2011, 76, 7229 CrossRef CAS PubMed.
- K. K. Rana, C. Guin and S. C. Roy, Tetrahedron Lett., 2000, 41, 9337 CrossRef CAS.
- K. K. Rana, C. Guin and S. C. Roy, Synlett, 2001, 1249 CrossRef CAS.
- S. C. Roy, K. K. Rana and C. Guin, J. Org. Chem., 2002, 67, 3242 CrossRef CAS PubMed.
- B. Banerjee and S. C. Roy, Synthesis, 2005, 2913 CAS.
- P. Chakraborty, S. Jana, S. Saha and S. C. Roy, Tetrahedron Lett., 2012, 53, 6584 CrossRef CAS PubMed.
- J. Justicia, L. Álvarez de Cienfuegos, A. G. Campaña, D. Miguel, V. Jakoby, A. Gansäuer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525 RSC.
- A. F. Barrero, A. Rosales, J. M. Cuerva and J. E. Oltra, Org. Lett., 2003, 5, 1935 CrossRef CAS PubMed.
- I. Abe, M. Rohmer and G. D. Prestwich, Chem. Rev., 1993, 93, 2189 CrossRef CAS.
- (a) D. L. J. Clive and S. R. Magnuson, Tetrahedron Lett., 1995, 36, 15 CrossRef CAS; (b) D. L. J. Clive, S. R. Magnuson;, H. W. Manning and D. L. Mayhew, J. Org. Chem., 1996, 61, 2095 CrossRef CAS; (c) L. Shi, K. Meyer and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49, 9250 CrossRef CAS PubMed.
- A. F. Barrero, J. M. Cuerva, M. M. Herrador and M. V. Valdivia, J. Org. Chem., 2001, 66, 407 CrossRef.
- M. Paradas, A. G. Campaña, T. Jiménez, R. Robles, J. E. Oltra, E. Buñuel, J. Justicia, D. J. Cárdenas and J. M. Cuerva, J. Am. Chem. Soc., 2010, 132, 12748 CrossRef CAS PubMed.
- K. Nakai, M. Kamoshita, T. Doi, H. Yamada and T. Takahashi, Tetrahedron Lett., 2001, 42, 7855 CrossRef CAS.
- A. F. Barrero, J. M. Cuerva, E. J. Álvarez-Manzaneda, J. E. Oltra and R. Chahboun, Tetrahedron Lett., 2002, 43, 2793 CrossRef CAS.
- A. F. Barrero, E. J. Álvarez-Manzaneda and R. Álvarez-Manzaneda, Tetrahedron Lett., 1989, 30, 3351 CrossRef CAS.
- J. F. Arteaga, V. Domingo, J. F. Quílez del Moral and A. F. Barrero, Org. Lett., 2008, 10, 1723 CrossRef CAS PubMed.
- (a) A. Gansäuer, D. Worgull and J. Justicia, Synthesis, 2006, 2151 CrossRef PubMed; (b) R. Tapia, M. J. Cano, H. Bouanou, E. Álvarez, R. Álvarez-Manzaneda, R. Chahboun and E. Álvarez-Manzaneda, Chem. Commun., 2013, 49, 10257 RSC.
- A. Rosales, J. Muñoz-Bascón, V. M. Morales-Alcázar, J. A. Castilla-Alcalá and J. E. Oltra, RSC Adv., 2012, 2, 12922 RSC.
- J. Justicia, J. E. Oltra, A. F. Barrero, A. Guadaño, A. González-Coloma and J. M. Cuerva, Eur. J. Org. Chem., 2005, 712 CrossRef CAS.
- (a) D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1 CAS , and earlier reviews cited therein; ; (b) J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2007, 24, 31 RSC , and reviews cited therein.
- (a) D. H. R. Barton, S. D. Bèviére and D. R. Hill, Tetrahedron, 1994, 50, 2665 CrossRef CAS; (b) N. Komiya, T. Naota and S. I. Murahashi, Tetrahedron Lett., 1996, 37, 1633 CrossRef CAS; (c) K. Chen, A. Eschenmoser and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 9705 CrossRef CAS PubMed; (d) M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed; (e) M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216 CrossRef CAS PubMed; (f) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed . For a recent review, see: ; (g) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed.
- T. Doi, S. Fuse, S. Miyamoto, K. Nakai, D. Sasuga and T. Takahashi, Chem.–Asian J., 2006, 1, 370 CrossRef CAS PubMed.
- A. F. Barrero, J. F. Quílez del Moral, M. M. Herrador, I. Loayza, E. M. Sánchez and J. F. Arteaga, Tetrahedron, 2006, 62, 5215 CrossRef CAS PubMed.
- (a) J. Justicia, J. E. Oltra and J. M. Cuerva, Tetrahedron Lett., 2004, 45, 4293 CrossRef CAS PubMed; (b) J. Justicia, J. E. Oltra and J. M. Cuerva, J. Org. Chem., 2005, 70, 8265 CrossRef CAS PubMed.
- (a) T. Jiménez, S. P. Morcillo, A. Martín-Lasanta, D. Collado-Sanz, D. J. Cárdenas, A. Gansäuer, J. Justicia and J. M. Cuerva, Chem.–Eur. J., 2012, 18, 12825 CrossRef PubMed; (b) J. A. González-Delgado, J. F. Arteaga, M. M. Herrador and A. F. Barrero, Org. Biomol. Chem., 2013, 11, 5404 RSC.
- (a) G. R. Pettit, Z. A. Cichacz, R. Tan, M. S. Hoard, N. Melody and R. K. Pettit, J. Nat. Prod., 1998, 61, 13 CrossRef CAS PubMed; (b) M. Cueto, P. R. Jensen and W. Fenical, Org. Lett., 2002, 4, 1583 CrossRef CAS PubMed.
- K. B. Sharpless, W. Amberg, Y. Bennani, G. A. Crispido, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768 CrossRef CAS.
- V. Domingo, L. Silva, H. R. Diéguez, J. F. Arteaga, J. F. Quílez del Moral and A. F. Barrero, J. Org. Chem., 2009, 74, 6151 CrossRef CAS PubMed.
- V. Domingo, J. F. Arteaga, J. L. López Pérez, R. Peláez, J. F. Quílez del Moral and A. F. Barrero, J. Org. Chem., 2012, 77, 341 CrossRef CAS PubMed.
- A. F. Barrero, M. M. Herrador, J. F. Quílez del Moral, P. Arteaga, J. F. Arteaga, H. R. Diéguez and E. M. Sánchez, J. Org. Chem., 2007, 72, 2988 CrossRef CAS PubMed.
- V. Domingo, H. R. Dieguez, C. P. Morales, J. F. Arteaga, J. F. Quílez del Moral and A. F. Barrero, Synthesis, 2010, 67 CAS.
- M. Yamaoka, A. Nakazaki and S. Kobayashi, Tetrahedron Lett., 2009, 50, 6764 CrossRef CAS PubMed.
- J. Justicia, A. G. Campaña, B. Bazdi, R. Robles, J. M. Cuerva and J. E. Oltra, Adv. Synth. Catal., 2008, 350, 571 CrossRef CAS.
- J. G. Urones, I. S. Marcos, P. Basabe, C. A. Alonso, D. Diez, N. M. Garrido, I. M. Oliva, J. S. Rodilla, A. M. Z. Slawin and D. J. Williams, Tetrahedron Lett., 1990, 31, 4501 CrossRef CAS.
- J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060 CrossRef CAS PubMed.
- (a) J. Justicia, J. L. Oller-López, A. G. Campaña, J. E. Oltra, J. M. Cuerva, E. Buñuel and D. J. Cárdenas, J. Am. Chem. Soc., 2005, 127, 14911 CrossRef CAS PubMed; (b) J. Justicia, T. Jiménez, D. Miguel, R. Contreras-Montoya, R. Chahboun, E. Álvarez-Manzaneda, D. Collado-Sanz, D. J. Cárdenas and J. M. Cuerva, Chem.–Eur. J., 2013, 19, 14484 CrossRef CAS PubMed.
- C. Pérez Morales, J. Catalán, V. Domingo, J. A. González Delgado, J. A. Dobado, M. Mar Herrador, J. F. Quílez del Moral and A. F. Barrero, J. Org. Chem., 2011, 76, 2494 CrossRef PubMed.
- (a) J. Justicia, L. Álvarez de Cienfuegos, R. E. Estévez, M. Paradas, A. M. Lasanta, J. L. Oller, A. Rosales, J. M. Cuerva and J. E. Oltra, Tetrahedron, 2008, 64, 11938 CrossRef CAS PubMed; (b) M. C. Pérez Morales, J. V. Catalán, V. Domingo, M. Jaraíz, M. M. Herrador, J. F. Quílez del Moral, J. L. López-Pérez and A. F. Barrero, Chem.–Eur. J., 2013, 19, 6598 CrossRef PubMed.
- (a) A. Fernández-Mateos, A. I. Ramos Silvo, R. Rubio González and M. S. J. Simmonds, Org. Biomol. Chem., 2012, 10, 5620 RSC; (b) A. Fernández-Mateos, S. Encinas Madrazo, P. Herrero Teijón, R. Rabanedo Clemente, R. Rubio González and F. Sanz González, J. Org. Chem., 2013, 78, 9571 CrossRef PubMed.
- (a) A. Gansäuer, D. Worgull, K. Knebel, I. Huth and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 8882 CrossRef PubMed; (b) A. Gansäuer, A. Greb, I. Huth, D. Worgull and K. Knebel, Tetrahedron, 2009, 65, 10791 CrossRef PubMed.
- (a) J. Y. Cha, J. T. S. Yeoman and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 14964 CrossRef CAS PubMed; (b) K. E. Lazarski, B. Akpinar and R. J. Thomson, Tetrahedron Lett., 2013, 54, 635 CrossRef CAS PubMed; (c) J. T. S. Yeoman, V. W. Mak and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 11764 CrossRef CAS PubMed.
- Y. Haruo, T. Hasegawa, H. Tanaka and T. Takahashi, Synlett, 2001, 1935 CrossRef CAS.
- B. M. Trost, H. C. Shen and J.-P. Surivet, Angew. Chem., Int. Ed., 2003, 42, 3943 CrossRef CAS PubMed.
- M. Oikawa, R. Hashimoto and M. Sasaki, Eur. J. Org. Chem., 2011, 538 CrossRef CAS.
- T. Yoshimitsu, S. Nojima, M. Hashimoto and T. Tanaka, Org. Lett., 2011, 13, 3698 CrossRef CAS PubMed.
- A. Odani, K. Ishihara, M. Ohtawa, H. Tomoda, S. Omura and T. Nagamitsu, Tetrahedron, 2011, 67, 8195 CrossRef CAS PubMed.
- S. Yamashita, N. Suda, Y. Hayashi and M. Hirama, Tetrahedron Lett., 2013, 54, 1389 CrossRef CAS PubMed.
- (a) J. Justicia, J. E. Oltra and J. M. Cuerva, J. Org. Chem., 2004, 69, 5803 CrossRef CAS PubMed; (b) A. Gansäuer, A. Rosales and J. Justicia, Synlett, 2006, 927 CrossRef PubMed; (c) A. Gansäuer, J. Justicia, A. Rosales, D. Worgull, B. Rinker, J. M. Cuerva and J. E. Oltra, Eur. J. Org. Chem., 2006, 4115 CrossRef.
- P. K. Mandal, G. Maiti and S. C. Roy, J. Org. Chem., 1998, 63, 2829 CrossRef CAS.
- P. K. Mandal and S. C. Roy, Tetrahedron, 1999, 55, 11395 CrossRef CAS.
- S. Saha and S. C. Roy, Tetrahedron, 2010, 66, 4278 CrossRef CAS PubMed.
- G. Ruano, M. Grande and J. Anaya, J. Org. Chem., 2002, 67, 8243 CrossRef CAS PubMed.
- G. Ruano, J. Martiáñez, M. Grande and J. Anaya, J. Org. Chem., 2003, 68, 2024 CrossRef CAS PubMed.
- (a) L. M. Monleón, M. Grande and J. Anaya, Tetrahedron, 2007, 63, 3017 CrossRef PubMed; (b) L. M. Monleón, M. Grande and J. Anaya, Synlett, 2007, 1243 Search PubMed; (c) L. M. Monleón, M. Grande and J. Anaya, Tetrahedron, 2012, 68, 10794 CrossRef PubMed.
- S. K. Mandal and S. C. Roy, Tetrahedron, 2008, 64, 11050 CrossRef CAS PubMed.
- T. K. Chakraborty, R. Samanta and K. Ravikumar, Tetrahedron Lett., 2007, 48, 6389 CrossRef CAS PubMed.
- L. Xu and X. Huang, Tetrahedron Lett., 2008, 49, 500 CrossRef CAS PubMed.
- J. L. Chiara, S. Bobo and E. Sesmilo, Synthesis, 2008, 3160 CrossRef CAS PubMed.
- M. Sreekanth, G. Pranitha, B. Jagadeesh and T. K. Chakraborty, Tetrahedron Lett., 2011, 52, 1709 CrossRef CAS PubMed.
- S. Basu, P. S. Kandiyal, R. S. Ampapathib and T. K. Chakraborty, RSC Adv., 2013, 3, 13630 RSC.
- S. Jana, C. Guin and S. C. Roy, Indian J. Chem., 2007, 45, 1648 Search PubMed.
- S. Saha, S. K. Mandal and S. C. Roy, Tetrahedron Lett., 2011, 52, 3128 CrossRef CAS PubMed.
- S. K. Mandal, M. Paira and S. C. Roy, J. Chem. Sci., 2010, 122B, 423 CrossRef PubMed.
- X. Zheng, X.-J. Dai, H.-Q. Yuan, C.-X. Ye, J. Ma and P.-Q. Huang, Angew. Chem., Int. Ed., 2013, 52, 3494 CrossRef CAS PubMed.
- G. Frey, H.-T. Luu, P. Bichovski, M. Feurer and J. Streuff, Angew. Chem., Int. Ed., 2013, 52, 7131 CrossRef CAS PubMed.
- J. Streuff, M. Feurer, P. Bichovski, G. Frey and U. Gellrich, Angew. Chem., Int. Ed., 2012, 51, 8661 CrossRef CAS PubMed.
- J. Velasco, X. Ariza, L. Badía, M. Bartra, R. Berenguer, J. Farràs, J. Gallardo, J. Garcia and Y. Gasanz, J. Org. Chem., 2013, 78, 5482 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |