Novel Pd based catalyst for the removal of organic and emerging contaminants†
Mallikarjuna N.
Nadagouda
*a,
Ishan
Desai
b,
Carlo
Cruz
a and
Duck J.
Yang
c
aThe U.S. Environmental Protection Agency, ORD, NRMRL, WSWRD, 26 W. Martin Luther King Dr Cincinnati, Ohio, 45268, USA. E-mail: Nadagouda.mallikarjuna@epa.gov; Tel: 01-513-569-7232
bEnvironmental and Water Resources Engineering Division, Zachry, Department of Civil Engineering, Texas A&M University, College Station, TX 77843, United States, USA
cDepartment of Chemistry, The University of Texas at Dallas, 800 W Campbell, BE26, Richardson, TX, 75080, USA. Tel: 972-883-6681
First published on 24th May 2012
Abstract
We report a simple approach to autocatalytic reduction and decoration of Au, Pd, and Pt nanocrystals on biodegradable cellulose polymer coated with polypyrrole. Cellulose was used as a template for the coating of polypyrrole. Reduction of different metals such as Au, Pd, and Pt and their decoration can be achieved through polypyrrole-coated cellulose fibers. Pre-selected bulk morphology of PdO/PdO2-embedded carbon was achieved through a microwave combustion method within one minute using the above-mentioned metal and polypyrrole-coated composite cellulose fibers. Synthesized Pd-decorated polypyrrole-coated cellulose fibers (Pd/py-cellulose) and microwave-ignited above mentioned (PdO/PdO2/C) materials provide a suitable catalyst for the degradation/reduction of methylene blue (waste water simulator) to leucomethylene blue at room temperature in aqueous media. At various concentrations, the material exhibits an overall reaction coefficient (ranging from 0.0183 to 0.1168 M−1 s−1), much greater than the self-degradation of methylene blue (MB) (0.00002 M−1 s−1). Through microwave ignition, the apparent increase in surface area of palladium results in a catalyst capable of high conversion (68.9%) at low concentrations.
Introduction
Bottom-up strategies for the synthesis of nanomaterials are integral components in nanoscience and nanotechnology. Lately, considerable interest has been focused on the possibility of construction of new assemblies from nanoparticles and other microcomponents to yield superstructured materials with unique and useful optical, electronic, and magnetic properties—properties that go beyond those of single elements.1–7 In search of biodegradable green polymers8,9 when nanoreinforced with nanoclay, nanotubes, and other nanostructures, create value-added applications of green polymers to support the next generation of materials possessing excellent properties.10–15 Among the renewable materials available, cellulose is the most abundant biopolymer provided by nature.16,17 Because of its availability and low cost, as well as its interesting chemical and physical properties, cellulose is widely used.18 Recently, research has focused on applications with high added-value, such as nanocomposites. These nanocomposites include cellulose nanocrystallites, e.g., nanocomposites of cellulose-based electrolytes,19 high performance composite materials,20 organoclay exfoliated cellulose with improved thermal properties,21 nanocomposite films with good tensile strength,22 biomedical applications,23 and nanocomposites that can be used as host materials.24 Consequently, present innovations are more focused on specific applications that result in decreased use of chemicals such as hydroxylamine hydrochloride and sodium borohydride, as there is renewed interest in green chemistry principles in materials design.25–31 Previously, Shin et al. reported the novel synthesis of titania,32 selenium,33 and gold–silver34 alloy nanoparticles on cellulose nanocrystals (CNXLs). Additionally, modified CNXLs have been shown to possess structure-directing capabilities.34 Similarly, Cai et al. produced various metal nanoparticles within a nanoporous cellulose gel matrix by hydrothermal or NaBH4 reduction.35 Moreover, the capabilities of CNXL as a reducing agent for platinum on carbon electrocatalysts has been extensively studied.36,37 Interestingly, Cirtiu et al. reports the use of monodispersed palladium nanoparticles on colloidal CNXL as a catalyst for hydrogenation and Heck coupling.38In this direction herein, we describe a simple green chemistry approach to autocatalytic reduction of Au, Pd and Pt crystals on polypyrrole-coated cellulose fibers. Instead of using the whole polypyrrole polymer fiber as the templating/reducing agent we have used cellulose fibers as a supporting matrix and a thin coating of polypyrrole polymer to act as the reducing agent. Through this process, at least 80% of nondegradable polymer (polypyrrole) can be avoided and this addresses several green chemistry principles. Additionally, polypyrrole synthesized in the absence of cellulose fibers forms spherical particles; consequently, the possibility of application in sensors and other technology is significantly reduced.
The improvement of facile, economical, and green methods for treatment of organic pollutants has been crucial in the field of environmental science and technology.39 Numerous industries use various dyes to achieve product coloration.40 Textile industries create enormous amounts of polluted effluents that are commonly discharged to surface water bodies and groundwater aquifers. These wastewaters damage the ecological structure of the receiving surface water and create disorder to the groundwater resources. Moreover, the majority of dyes used in textile industries are stable to light and are not biodegradable. In order to condense the risk of environmental pollution from such wastes, it is obligatory to treat them prior to discharging to the receiving environments. Accordingly, they produce a substantial quantity of colored wastewater. The efficacy of the reduction/adsorption for dye from wastewater has made it an ideal alternative to other expensive treatment methods.41 Methylene blue (MB) is a water-soluble cationic dye molecule that has been widely studied and has been used as simulated wastewater.41 Different methods have been attempted for the reduction/degradation of methylene blue.42–44 Reduction of methylene blue to leucomethylene blue on illuminated rutile crystals in methanolic and aqueous solutions containing hydrogen chloride was investigated colorimetrically and electrochemically.42 Methylene blue reduction by ascorbic acid was achieved by Ogren et al.43 Fujishima et al. employed UV-Irradiated TiO2 for bleaching and degradation of methylene blue.44 Herein we report the autocatalytic synthesis of Au, Pt and Pd decorated polypyrrole-coated cellulose fiber with Pd-decorated fibers exhibiting potential for MB reduction. Additionally, microwave ignition of the aforementioned composite materials results in a pre-selected bulk morphology, and a suitable catalyst for the reduction of methylene blue to leucomethylene blue at room temperature in aqueous media.
A wide variety of emerging contaminants have been recently found in wastewater effluents, which are of concern because of their adverse effects on human health and the environment. Detergents and detergent metabolites (including alkylphenols (APs), alkylphenol ethoxylates (APEOs) and alkylphenol carboxylates (APECs)), antimicrobial agents (triclosan) and heribicides (Atrazine) are some of the widely occurring contaminants among them. Octylphenol (OP) is the degradation product of the octylphenol ethoxylates. OPs have been shown to have the ability to bind with estrogen receptors and alter the way it functions. Atrazine (ATR) is a recalcitrant pollutant in the environment due to its very low biodegradation. Triclosan (TCS) is a potential endocrine disruptor and have been shown to cause toxicity in aquatic organisms. Because these chemicals are commonly present in treated wastewater effluent and are discharged into the aquatic environment, research exploring technologies used to remove these chemicals from wastewater is of interest for utilities and public stakeholders. In this paper we have evaluated the reactivity of Pd based catalyst with OP, ATR and TCS. The objective of this work is to show the versatility of the prepared catalyst in treating different contaminants present in wastewater.
Results and discussion
Formation of polypyrrole-coated cellulose fibers was achieved through oxidative polymerization of pyrrole in the presence of cellulose using 1 N FeCl3 as an oxidant. In the initial steps of the polymerization reaction, the dispersed cellulose fibers in the water adsorb the added pyrrole monomer and then, by addition of an oxidant, polymerization is initiated. The solution turns to dark green, indicating polypyrrole formation on the surface of cellulose fibers. Polypyrrole is an organic polymer with very high conductivity and good reduction potential to reduce metals.38 Also, it is capable of forming films on various surfaces including glass. The obtained polypyrrole-coated cellulose fibers were used to reduce different metals namely gold, platinum, and palladium. The XRD pattern of polypyrrole-coated cellulose fibers is shown in ESI Fig. S1.† Major peaks were indexed to the cellulose polymer (JCPDS pattern number 00-50-2241). Polypyrrole can form an amorphous phase with a broad peak around 2θ 20–30°.39 In the present case, cellulose diffraction peaks overlap with polypyrrole peaks and it is difficult to index separately.Noble metals have different reduction potentials, for example, the reduction potential of Pt 1.20 V, Pd 0.915 V and Au 1.50. As-synthesized polypyrrole-coated cellulose fibers can spontaneously reduce noble metal ions to the corresponding metal nanostructures at room temperature without the use of any added capping or dispersing agents. For example, Au, Pt, and Pd, etc., can be reduced and deposited readily on the surface of polypyrrole-coated cellulose fibers. The Au3+/Au system takes a greater amount of time for the reduction compared to Pd.
Spherical particles were formed in case of Au (ESI,† Fig. S2 (a and b)) and the same trend was continued for Pd and Pt (Fig. 1 (a–d)). The increase in polypyrrole (1.441 M of pyrrole) coating had no influence on shapes of noble metal crystals. Au, Pt and Pd retained their shapes and structures with an increase in surface layer assembly thickness (see ESI,† Fig. S3 (a and b)), Fig. 2 (a–d) and ESI,† Fig. S4. Further increase in polypyrrole content (10.0 mL pyrrole) on cellulose fibers yielded very thick, self-assembled layers of noble metal crystals (see Fig. 3 (a–d) and 4 (a–d)) and ESI,† Fig. S5). We have extended this strategy to decorate Fe nanostructures on cellulose nanofibers (ESI,† Fig. S6).
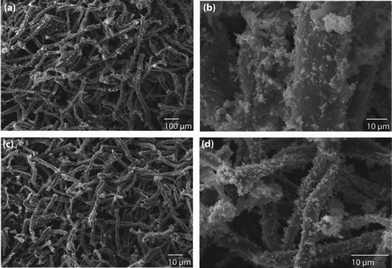 | ||
| Fig. 1 The decoration of (a and b) Pd and (c and d) Pt nanoparticles on polypyrrole-coated cellulose fibers, prepared using a low concentration of pyrrole (0.2882 M). | ||
 | ||
| Fig. 2 The decoration of (a and b) Pd and (c and d) Pt nanoparticles on polypyrrole-coated cellulose fibers, prepared using a medium concentration of pyrrole (1.441 M). | ||
 | ||
| Fig. 3 The decoration of (a and b) Au nanoparticles on polypyrrole-coated cellulose fibers, prepared using a high concentration of pyrrole (2.882 M). | ||
 | ||
| Fig. 4 The decoration of (a and b) Pd and (c and d) Pt nanoparticles on polypyrrole-coated cellulose fibers, prepared using a high concentration of pyrrole (2.882 M). | ||
XRD was used for phase confirmation of decorated noble metal crystals on polypyrrole-coated cellulose fibers and are shown in the Fig. S6–S8 (ESI†) for Au, Pd, and Pt respectively. All the patterns were compared with JCPDS patterns and corresponding pattern numbers are given in the XRD patterns. Au and Pd were crystalline in nature, whereas Pt crystals did not show any diffractable peaks. The low concentration of Pt, which was not within the detectable limit of XRD, cannot be dismissed. We extended this study to reduce Fe nanoparticles and found that there was a significant difference in morphology and crystallinity.
ESI,† Fig. S6 shows the SEM images of self assembled iron particles on polypyrrole-coated cellulose fibers. Irrespective of polypyrrole thickness, a very thick coating of an iron layer was observed. The XRD pattern (see ESI,† Fig. S10) indicated that the Fe nanoparticles formed were amorphous in nature.
X-Ray mapping was used for an additional confirmation of the formation of noble nanostructures on polypyrrole-coated cellulose fibers and is shown in Fig. 5. The energy dispersive X-ray analyses (EDS) analyses was completed on various samples and are given in the ESI,† Table S1 to S3.
 | ||
| Fig. 5 X-Ray mapping images of (a) Au (b) Pt, and (c) Pd nanostructures on polypyrrole-coated cellulose fibers. (Respective colour dots indicate presence of metal/metal salt.) | ||
TEM images of autocatalytic reduced Pd nanoparticles on 1.441 M polypyrrole-coated cellulose fibers are shown in Fig. 6. Cubes with sizes ranging from 80–100 nm were observed. A primary factor that determines the habit of a crystal is the relative rates at which different crystal planes grow. Their surrounding environment also plays a part. Pd normally reduces quickly as compared to Au and Pt due to lower reduction potential.
 | ||
| Fig. 6 TEM image of autocatalytic-reduced Pd nanoparticles on polypyrrole (1.441 M)-coated cellulose fibers. | ||
An X-ray photoemission spectrum of Au nanoparticle decorated polypyrrole-coated cellulose fibers prepared using a medium concentration of pyrrole (1.441 M) is shown in Fig. 7. Usually, the peaks at 84.0 and 87.7 eV indicate metallic gold and the peaks at 85.8 and 89.8 eV indicate oxidized Au (Au+3) in the XPS analysis.45-48 Also, the presence of peaks at 85.6–85.8 eV represents a Au+1 state. From the figure, two peaks at 84.2 eV and 87.8 eV indicate the binding energy of bulk metallic gold. Fig. 8 shows four main peaks at 336.5, 339.4, 341.8, 344.3 eV for XPS analysis of Pd. Two peaks at 341.8 and 336.5 eV indicate the binding energy of 3d3/2 and 3d5/2 of PdO, respectively. A peak at 339.4 eV was observed, which indicates the binding energy of metallic Pd (3d3/2), however, the main peak of Pd 3d5/2 at 334.6 eV was not observed. Usually, two peaks at 334.6 and 340.1 eV indicate Pd metal and the peak at 334.6 eV is mainly used to indicate Pd metal. Therefore, the peak at 339.4 eV did not indicate metallic Pd in this sample. In this case, the peaks at 339.4 and 344.3 eV might indicate 3d5/2 and 3d3/2 of PdO2.49–54 However, the XRD patterns indicate bulk Pd metal formation. The probable explanation is that the surface must have oxidized and a majority of the core retains metal.
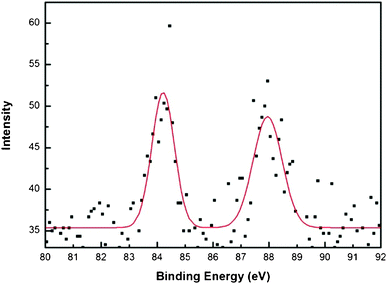 | ||
| Fig. 7 X-Ray photoemission spectrum of Au nanoparticle decorated polypyrrole-coated cellulose fibers prepared using a medium concentration of pyrrole (1.441 M). | ||
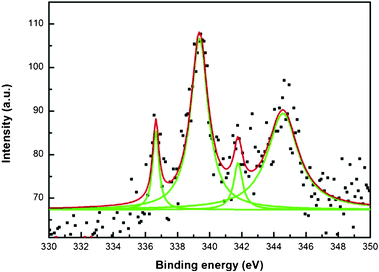 | ||
| Fig. 8 X-Ray photoemission spectra of a Pd nanoparticle decorated polypyrrole-coated cellulose fibers prepared using a medium concentration of pyrrole (1.441 M). | ||
Methylene blue (simulating color waste water dye) exhibits a visible spectrum at 665 nm (see Fig. 9a). Upon addition of the Pd/pyrrole-cellulose catalyst, the loss of color in the stock solution is apparent even at low concentrations of the catalyst (Fig. 9b–d). Increased concentrations of the catalyst elucidate higher potentials for color removal (reduction/degradation), showing a significant absorbance difference after 120 min. The formation mechanism is shown in Scheme 1. Partial adsorption of methylene blue on polypyrrole-coated cellulose fibers cannot be ruled out.
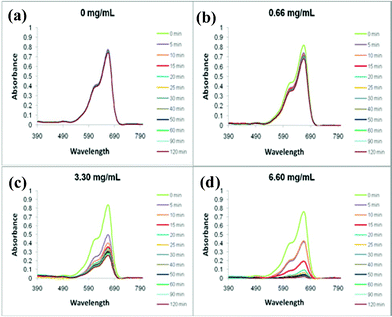 | ||
| Fig. 9 The UV-Visible spectra of various amounts (a. 0 mg mL−1; b. 0.66 mg mL−1; c. 3.30 mg mL−1; d. 6.60 mg mL−1) of Pd/pyrrole-cellulose throughout standard reaction conditions. | ||
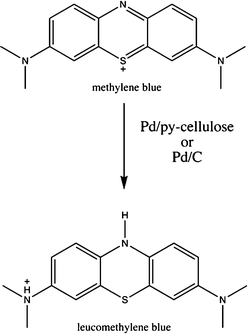 | ||
| Scheme 1 Formation mechanism of colorless leucomethylene blue. | ||
The pre-selected morphology of Pd-embedded carbon (PdO/PdO2/C) can be obtained when polypyrrole-coated cellulose fibers are heated in a conventional microwave oven. The temperature rises to red hot in a few seconds resulting in a rapid loss of heteroatoms, dopant ions, cellulose etc. to yield a self-retained, pre-selected morphology of carbon. Similar morphologies were observed in conducting polymers.55 The bulk morphology of the composite is retained after microwave heating except in the chemical composition. By weight, 21.58% of the catalyst is recovered following microwave ignition (78.42% weight loss). The formation mechanism of pre-selected Pd coated carbon is similar to nanocarbons obtained from conducting polymer polypyrrole.55 Electronic conductivity of the polymer helps, i.e. conducting ions aligning with microwaves. In the process of conducting ion alignment, a great deal of heat is generated and is sufficient enough to ignite the cellulose fibers. Hydroxyl functional groups from the cellulose can also couple with microwave irradiation to self-propagate and ignite the cellulose material. The SEM image is shown in Fig. 10. The particle sizes (measured by DLS, Malvern Nanoseries) are in the range of 1.5 μm as they are tied together to make rod shaped structures (see Fig. 10 for SEM image). All other samples were beyond the detectable limit of the instrument.
 | ||
| Fig. 10 SEM image of microwave-ignited Pd coated carbon fibers. | ||
Fig. 11 shows four main peaks at 336.6, 338.6, 341.9, and 344.1 eV for XPS analysis of pre-selected morphology of PdO/PdO2-embedded carbon. Two peaks at 341.9 and 336.6 eV indicate the binding energy of 3d3/2 and 3d5/2 of PdO, respectively. The other two peaks at 338.6 and 344.1 indicate the binding energy of oxidized PdO2.49–54 The XPS spectra clearly indicate the oxidation of Pd to PdO and PdO2. The main reason for this is due to oxidation of Pd while it is combusted in the presence of microwaves. Also, metals react vigorously in the presence of microwaves.
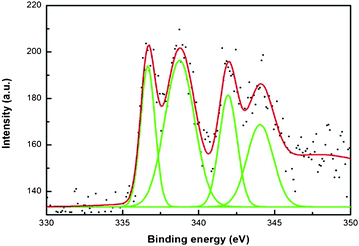 | ||
| Fig. 11 X-Ray photoemission spectra of Pd nanoparticle decorated polypyrrole-coated cellulose fibers, prepared using a medium concentration of pyrrole (1.441 M) and ignited under microwave irradiation for 1 min. | ||
The obtained microwave-ignited, pre-selected PdO/PdO2 embedded carbon was used for the degradation of methylene blue dye and the absorption spectra are shown in Fig. 12.
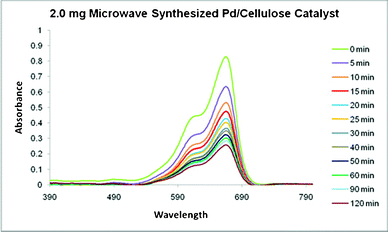 | ||
| Fig. 12 The absorption spectra of methylene blue in the presence of microwave-ignited Pd/polypyrrole-cellulose over time. | ||
The absorption of methylene blue was analyzed at 665 nm, and the overall reaction rate constant was determined. [MB∞] was estimated by mathematical modeling of experimental values of [MB]. The linear characteristics of [MB-MB∞]−1versus time suggest the degradation of MB follows second order kinetics (Fig. 13). Previous reports conclude similar kinetics in aqueous systems. The overall reaction rate constants, k, for 2.0 mg, 10.0 mg, and 20.0 mg Pd/py-cellulose were found to be 0.1168, 0.0319, and 0.0183 M−1 s−1, respectively.
![The linear nature of [MB-MB∞]−1 over 120 min. The correlation coefficient of the corresponding linear regression is presented adjacent to the respective graphs.](/image/article/2012/RA/c2ra20562a/c2ra20562a-f13.gif) | ||
| Fig. 13 The linear nature of [MB-MB∞]−1 over 120 min. The correlation coefficient of the corresponding linear regression is presented adjacent to the respective graphs. | ||
Here, k notably decreases with increasing catalyst mass (Fig. 13). However, the conversion of MB with increasing catalyst concentration exhibits the opposite trend (Fig. 14). The amount of catalyst present appreciably impacts the total conversion of MB in the studied reaction. Although greater conversion is achieved with larger masses, the reaction exhibits a much lower overall rate. Yet, 2.0 mg Pd/polypyrrole-cellulose exhibits a much slower initial instantaneous loss of MB at −0.2494 M s−1 than 20.0 mg Pd/py-cellulose (−1.0916 M s−1). Additionally, the microwave-synthesized Pd/C structures exhibit considerable conversion with a low k value. Moreover, the initial instantaneous rate of these structures is also greater than its mass equivalent Pd/py-cellulose at −0.6088 M s−1. The BET surface area of the control sample is 12.1575 m2 g−1. After microwave ignition the surface increased to 60.6141 m2 g−1. Here, the increased surface area allows for a greater number of reaction collisions; consequently, the overall conversion of MB is affected.
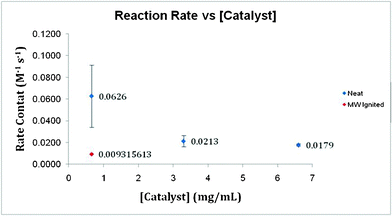 | ||
| Fig. 14 A comparison of the overall reaction rate k with varying catalyst concentration. Additionally, microwave-ignited catalyst k value is presented. | ||
At 0.66 mg mL−1 control cellulose-polypyrrole fibers, only 1.66% of the initial methylene blue is removed over a 120 min time period. At 6.60 mg mL−1 control cellulose-polypyrrole, only 18.27% of the initial methylene blue is removed over a 120 min time period. Comparatively, the catalytic Pd composite shows removal of 12% and 87% at 0.66 and 6.60 mg mL−1, respectively.
The catalyzed redox reaction of small metal particles proceeds through an electrochemical mechanism where electron transfer occurs via the metal particles. The electrochemical potential of the particles should be intermediate between the electron donor and acceptor. The reduction potential of the Pd(II)/Pd metal (aqueous) system is +0.987 V (vs. NHE) which becomes lower for Pd(II)/Pd nanoparticle systems and depends on particle size and the nature of the adsorbed ions at the particle surface. Small palladium particles act as electron transfer catalysts. Catalytic rates depend on the E1/2 of both the dye and Pd/PdO2 nanoparticles. Methylene blue has the reduction potential of 0.53 V in its oxidized form. We believe Pd(II)/Pd metal (aqueous) systems should be able to reduce the methylene blue due to a reduction potential difference. Also, catalytic activity of Pd/PdO2 cannot be ruled out.
In order to demonstrate the versatility of the catalyst material, degradation of octylphenol, triclosan and atrazine was undertaken and data are shown in Fig. 15–17. Octylphenol was removed by 56% in the first 20 min of the reaction. Within the time frame studied in our experiments (100 min) the overall removal of octylphenol was 72% (see Fig. 15). Almost 70% of the triclosan was removed in the first 20 min and the reaction slowed thereafter giving an overall removal of 79% in 100 min (Fig. 16). Atrazine was more recalcitrant of all the three organics. There was an overall reduction of 17% in atrazine in 100 min (Fig. 17). Each of the reaction kinetics shows a similar trend where there is faster kinetics in the initial times and the rate of reaction decreases as the reaction proceeds. This could be due to a limited number of active sites present on the surface of the catalyst or the surface catalyst changing to a less active structure as the reaction progresses. We are currently evaluating the various factors like organic dosage, catalyst dosage, pH, temperature, stirring speed on the reaction rates and the results would be presented in a follow-up paper. Also the partial adsorption cannot be ruled out. Before the oxidation, the bare Pd particles did not show any appreciable results for the organic and emerging contaminants.
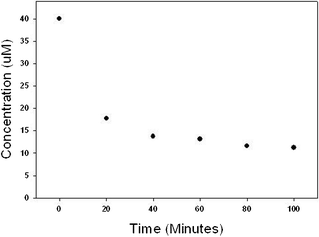 | ||
| Fig. 15 Removal of octylphenol using PdO/PdO2 catalyst. Reaction contained 40 μM octylphenol and 2 g L−1 of catalyst initially. | ||
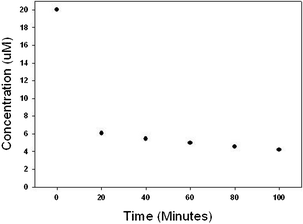 | ||
| Fig. 16 Removal of triclosan using PdO/PdO2 catalyst. Reaction contained 20 μM triclosan and 2 g L−1 of catalyst initially. | ||
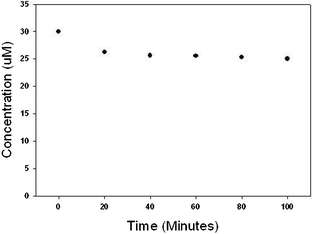 | ||
| Fig. 17 Removal of atrazine using PdO/PdO2 catalyst. Reaction contained 30 μM atrazine and 2 g L−1 of catalyst initially. | ||
Conclusion
Formation and decoration of noble crystals such as Au, Pd and Pt on polypyrrole-coated cellulose fibers was achieved through a greener approach reducing the usage of complete polypyrrole. Depending upon the polypyrrole thickness, the formation of different shapes, sizes, structures and decoration of metal crystals were observed. Different shapes such as cubes, prisms, and spherical structures were formed on polypyrrole-coated cellulose fibers. XRD revealed the phase formation. The pre-selected morphology of PdO/PdO2-embedded carbon was achieved using the microwave combustion method within 1 min. The aforementioned synthesized Pd/polypyrrole-cellulose and microwave-ignited PdO/PdO2/C materials provide a suitable catalyst for the reduction/degradation of methylene blue to leucomethylene blue at room temperature in aqueous media. At various concentrations, the material exhibits an overall reaction coefficient (ranging from 0.0183 to 0.1168 M−1 s−1) that is much greater than the self-degradation of MB (0.00002 M−1 s−1). Through microwave ignition, the apparent increase in surface area of PdO/PdO2/C results in a catalyst capable of high conversion (68.9%) at low concentrations. These self-assembled structures with noble crystals may find wide variety applications such as catalysis, fuel cell, membranes, and medicinal applications (micro actuator, drug carrying membrane, etc).Experimental section
Materials and methods
Methylene blue reduction
A stock solution of methylene blue (MB) was prepared by dissolving a known amount of MB (0.016 g, 0.5 mmol) into 40 mL distilled water. 30.0 μL stock solution was diluted by 3.0 mL distilled water and absorbance at 665 nm was measured by Cary 50 UV-Vis Spectrophotometer. Subsequently, a pre-determined amount of catalyst (0 to 20 mg) was added and spectrophotometric measurements were taken in five-minute intervals for thirty minutes. Following the initial rapid sampling period, measurements were taken at 40, 50, 60, 90, and 120 min after addition of catalyst. MB concentration throughout the reaction was determined by UV-Vis spectrophotometric methods.Degradation of octylphenol, triclosan and atrazine
The stock solutions of octylphenol (4 mM), triclosan (1 mM) and atrazine (3 mM) were each prepared in methanol. For the kinetic experiments, a 2 g L−1 dosage of catalyst was added into ultrapure water in a 50 mL glass vial and allowed to mix properly on a rotator for 30 min. The reaction was initiated by the addition of the stock organics to get the desired concentration and the reactor was subsequently kept on a rotator. The initial concentration for octylphenol, triclosan and atrazine was maintained at 40 μM, 20 μM and 30 μM respectively. At each time point a sample was drawn from the vial and centrifuged immediately. The supernatant was collected for organic analysis.The organics in reaction solutions was determined using a direct injection in a HPLC (Agilent 1200 series) equipped with a diode-array UV detector. Chomatography was performed using a C18 column (3 μm, 2.1 mm × 100 mm) at a flow rate of 0.5 mL min−1 under isocratic conditions. The mobile phase used in our analysis was a mixture of acetonitrile and water containing 0.1% glacial acetic acid. The ratio of acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reagent water containing 0.1% glacial acetic acid for triclosan, octylphenol and atrazine was maintained at 60:30, 50:50 & 30:70 respectively.
:reagent water containing 0.1% glacial acetic acid for triclosan, octylphenol and atrazine was maintained at 60:30, 50:50 & 30:70 respectively.
Microwave synthesis of pre-selected PdO/PdO2 embedded carbon fibers
About 20 mg of 1.441 M polypyrrole-coated cellulose fibers reduced with Pd was placed in a glass vial. Subsequently, the material was subjected to 800 W kitchen microwave irradiation for 1 min. Initially, sparks evolve from the fibers, and after continuous heating, the material emits a white flame. The contents were cooled to room temperature and analyzed by scanning electron microscopy (SEM) and energy dispersive X-ray analysis (EDX).X-Ray diffraction studies (XRD)
XRD was used to identify crystalline phases of solids. PANalytical Xpert Pro theta-two theta diffractometer using a Cu-Kα radiation at 45 kV and 40 mA was used. Scans were typically over the range 5 to 90 degrees (2 theta). A pattern analysis was performed using Jade+ software, v.7 or later (MDI, Inc., Livermore, CA), which generally followed the ASTM D934-80 procedure. Reference patterns were from the 2002 PDF-2 release from the ICDD (International Center for Diffraction Data, Newtown Square, PA).Morphology
For SEM, a JEOL-6490LV with an Oxford X-Act EDS system was used for imaging and elemental analysis. Images and EDS spectra were captured using an accelerating voltage of 30 kV. Spectra were collected for 60 live seconds using a process time of 30% and a 40% dead-time.BET surface area analysis
A Tristar 3000 (Micromeritics) porosimeter analyzer was used to determine BET surface area, pore volume, BJH pore size and pore size distribution.X-Ray photoelectron analysis (XPS)
XPS analysis was done using Kratos Axis Ultra Imaging X-ray photoelectron spectrometer at TAMU Materials Characterization Facility.Acknowledgements
The authors would like to thank Emily Nauman of Pegasus Corporation and John Olszewski for assistance with manuscript review and editing. Use of the TAMU Materials Characterization Facility and Dr Jing Wu are acknowledged.References
- M. Alexander, C. Xiao, B. Sinan, K. Mato, K. Anan and K. Klaus, Eur. J. Inorg. Chem., 2005, 18, 3717 Search PubMed.
- E. Susie and M. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- M. N. Nadagouda, C. Bennett-Stamper, C. White and D. Lytle, RSC Adv., 2012, 2(10), 4198–4204 RSC.
- P. Jain, X. Huang, I. El-sayed and M. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS.
- J. Dahl, B. Maddux and J. Hutchison, Chem. Rev., 2007, 107, 2228 CrossRef CAS.
- M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- C. Love, L. Estroff, J. Kriebel, R. Nuzzo and G. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef.
- M. N. Nadagouda and R. S. Varma, Acc. Chem. Res., 2011, 44, 469 CrossRef CAS.
- H. Park, M. Misra, L. Drzal and A. Mohanty, Biomacromol., 2004, 5, 281 Search PubMed.
- R. Gross and B. Kalra, Science, 2002, 297, 803 CrossRef CAS.
- H. Park, X. Liang, A. Mohanty, M. Misra and L. Drzal, Macromolecules, 2004, 37, 9076 CrossRef CAS.
- S. S. Ray and M. Bousmina, Prog. Mater. Sci., 2005, 50, 962–1079 CrossRef CAS.
- M. Lima and R. Borsali, Macromol. Rapid Commun., 2004, 25, 771 CrossRef.
- M. Samir, F. Alloin and A. Dufresne, Biomacromolecules, 2005, 6, 612 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Macromol. Rapid Commun., 2007, 28, 465 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Macromol. Rapid Commun., 2008, 29, 155–159 CrossRef CAS.
- F. Bovey, E. Winslow, “An Introduction to Polymer Science”Academic Press, New York, 1981, 518 Search PubMed.
- H. Krassig, R. Steadman, K. Schliefer, W. Albrecht, “Ullmann's Encyclopedia of Organic Compounds” 5th edn, VCH, New York, 1986, A5, p. 325 Search PubMed.
- E. Cranston and D. Gray, Biomacromolecules, 2006, 7, 2522 CrossRef CAS.
- R. Kohler and K. Nebel, Macromol. Symp., 2006, 244, 97 CrossRef CAS.
- H. Park, X. Liang, A. Mohanty, M. Misra and L. Drzal, Macromolecules, 2004, 37, 9076 CrossRef CAS.
- D. Ruan, L. Zhang, Z. Zhang and X. Xia, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 367 CrossRef CAS.
- W. Czaja, D. Young, M. Kawecki and R. Brown Jr., Biomacromolecules, 2007, 8, 1 CrossRef CAS.
- D. Ruan, Q. Huang and L. Zhang, Macromol. Mater. Eng., 2005, 290, 1017–1024 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2006, 8, 516 RSC.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2007, 9, 632 RSC.
- M. N. Nadagouda and R. S. Varma, Biomacromolecules, 2007, 8, 2762 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Cryst. Growth Des., 2007, 7, 2582 CAS.
- P. Anastas, J. Warner, Green Chemistry: Theory and Practice; Oxford University Press Inc.: New York, 1998 Search PubMed.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2008, 10, 859 RSC.
- M. N. Nadagouda, V. Polshettiwar and R. S. Varma, J. Mater. Chem., 2009, 19, 2026–2031 RSC.
- Y. Shin and G. Exharhos, Mater. Lett., 2007, 61, 2594 CrossRef CAS.
- Y. Shin, J. Blackwood, I. Bae, B. Arey and G. Exarhos, Mater. Lett., 2007, 61, 4297 CrossRef CAS.
- Y. Shin, I. Bae, B. Arey and G. Exarhos, J. Phys. Chem. C, 2008, 112, 4844 CAS.
- J. Cai, S. Kimura, M. Wada and S. Kuga, Biomacromolecules, 2009, 10, 87 CrossRef CAS.
- K. Benaissi, L. Johnson, D. Walsh and W. Thielemans, Green Chem., 2010, 12, 220 RSC.
- L. Johnson, W. Thielemans and D. Walsh, Green Chem., 2011, 13, 1686 RSC.
- C. Cirtiu, A. Dunlop-Biere and A. Moores, Green Chem., 2011, 13, 288 RSC.
- K. Wang, L. Yu, S. Yin, H. Li and H. Li, Pure Appl. Chem., 2009, 81, 2327 CrossRef CAS.
- S. Saad, S. Daud, F. Kasim and M. Saleh, ICOSM, 2007, 293 Search PubMed.
- R. Tayade, T. Natarajan and H. Bajaj, Ind. Eng. Chem. Res., 2009, 48, 10262 CrossRef CAS.
- H. Yoneyama, Y. Toyoguchi and H. Tamura, J. Phys. Chem., 1972, 76, 3460 CrossRef CAS.
- S. Mowry and P. Ogren, J. Chem. Educ., 1999, 76, 970 CrossRef CAS.
- T. Tatsuma, S. Tachibana, T. Miwa, D. Tryk and A. Fujishima, J. Phys. Chem. B, 1999, 103, 8033 CrossRef CAS.
- E. Irissou, M.C. Denis, M. Chaker and D. Guay, Thin Solid Films, 2005, 472, 49 CrossRef CAS.
- J. J. Pireaux, M. Liehr, P. A. Thiry, J. P. Delrue and R. Caudano, Surf. Sci., 1984, 141, 221 CrossRef CAS.
- L. K. Ono and B. R. Cuenya, J. Phys. Chem. C, 2008, 112, 4676 CAS.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215 CrossRef CAS.
- I. B. Rufus, V. Ramakrishnan, J. C. Viswanathan and J. C. Kuriacose, J. Mater. Sci. Lett., 1993, 12, 1536 CAS.
- D. Zemlyanov, B. Aszalos-Kiss, E. Kleimenov, D. Teshner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, H. Gabasch, W. Unterberger, K. Hayek and B. Klötzer, Surf. Sci., 2006, 600, 983 CrossRef CAS.
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125 CrossRef CAS.
- L. Hilaire, P. Legare, Y. Holl and G. Maire, Solid St. Commun., 1974, 32, 197 Search PubMed.
- K. S. Kim, A. F. Gossman and N. Winogard, Anal. Chem., 1974, 46, 197 CrossRef CAS.
- T. H. Fleisch and G. J. Mains, J. Phys. Chem., 1986, 90, 5317 CrossRef CAS.
- X. Zhang and S. Manohar, J. Am. Chem. Soc., 2005, 127, 1415 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional SEM images, BET surface area, XRD, and EDX data are available. See DOI: 10.1039/c2ra20562a/ |
| This journal is © The Royal Society of Chemistry 2012 |