Fuzziness: linking regulation to protein dynamics†
Monika
Fuxreiter
ab
aInstitute of Enzymology, Biological Research Center, Hungarian Academy of Sciences, 1518 Budapest, PO Box 7, Budapest, Hungary. E-mail: monika@enzim.hu
bMedical Research Council, Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH, UK
First published on 19th September 2011
Abstract
Proteins are dynamic creatures. Intrinsically disordered proteins (IDPs) function as multiplicity of structures and their activities can only be described by stochastic structure–function relationships. In their complex forms, however, IDPs were thought to lose their plasticity and behave similarly to globular proteins. Although various IDPs indeed fold upon binding, this view is not valid in general. IDPs usually interact with their partners via short motifs, which require malleable environments to function. Consequently, segments of IDPs could retain their disordered state in the complex, a phenomenon termed as fuzziness. Since its recognition, the number of structurally characterized fuzzy complexes, both with protein and DNA, rapidly increases. Here I review recent advances in our understanding of fuzziness. Four basic mechanisms are described how conformationally heterogeneous regions impact specificity or binding affinity of protein complexes. A novel allostery-model is proposed, where the regulatory site modulates the conformational equilibrium of the binding interface without adopting a unique structure. Protein–protein interactions, post-translational modifications or alternative splicing of the highly flexible/disordered regions offer further opportunities for regulation and expand the functional repertoire of fuzzy complexes.
 Monika Fuxreiter | Monika Fuxreiter received her degrees from the Eötvös Loránd University, Budapest, Hungary. She is a senior scientist at the Institute of Enzymology, Hungarian Academy of Sciences in Budapest, Hungary and a visiting scientist at the Laboratory of Molecular Biology, Medical Research Council, Cambridge. Her main interest is to analyze recognition mechanisms of intrinsically disordered proteins and develop models for fuzzy complexes. She aims to establish a stochastic structure–function relationship for proteins. She has two children and is a recipient of the L'Oreal-Unesco Women in Science prize, in Hungary. |
Introduction
Dynamics is inherently related to protein functionality. Although activity of many proteins is engraved in a stable three-dimensional architecture, it cannot be achieved without motions on different timescales ranging from atomic fluctuations to conformational rearrangements.1 Enzymatic catalysis, for example, is under strong influence of μs–ms motions that modulate the accessibility and optimal environment of the active site.2–4 Allosteric communication is realized via dynamical networks,5 formed by a set of physically connected residues.6 Several pieces of evidence demonstrate that function-related collective motions are conserved in orthologous protein families.7 All these motions, however, can be considered as variations around an optimal structure and could still be interpreted within the framework of the classical structure–function paradigm.Dynamics results in conformational ensembles, and indeed many proteins exist simultaneously in different, yet functionally relevant conformations.8 These proteins that can only be characterized by multiple structures are termed as intrinsically disordered proteins (IDPs).9 Although initially the concept was recognized with skepticism, it has been widely accepted by now. IDPs created a field on their own, where dynamics is of extraordinary importance. The role of IDPs is underscored by their preponderance in eukaryotic proteomes, enabling various complex functions.10 The plasticity of IDPs is exploited in regulatory pathways, where IDPs interact with other proteins, nucleic acids or small molecules.11 Upon binding their partners however, IDPs were assumed to lose most of their conformational freedom and adopt a well-defined structure.12 Therefore IDPs in complexes were thought to lack conformational ambiguity, which they possessed in unbound form.
Why do we raise such a boundary for protein dynamics? Abandoning the benefits of the structural variety and the ease of adaptability of IDPs does not seem to be an optimal solution in complexes, which need to be assembled or disassembled in a tightly controlled manner upon responding to diverse cellular cues. Indeed, the conformational ambiguity of IDPs is preserved in a variety of complexes.13–15 This phenomenon, which is termed fuzziness, polymorphism or actual structural disorder not only exists, but also serves key functional roles. Four structural categories of fuzzy complexes were defined, regarding the type and location of the intrinsically disordered (ID) segment (Box 1, Fig. 1).
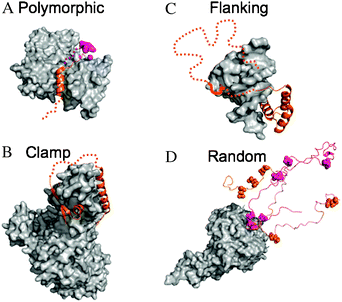 | ||
| Fig. 1 Examples of different structural categories of fuzzy complexes. The IDPs are shown by orange and magenta, fuzzy regions are represented by dotted lines. The binding partners are displayed as gray surfaces. (A) Polymorphic complex: the WH2 domain of Wiskott–Aldrich syndrome protein interacts with actin in alternative locations: via an 18 residue segment (orange; PDB code 2a3z) or via only 3 residues (magenta; PDB code 2ff3). (B) Clamp complex: the nonsense mediated decay factor UPF2 binds to UPF1 via two structured regions (PDB code 2wjv) and the connecting linker remains ambiguous in the complex (dotted line). (C) Flanking complex: DNA-binding by the transcription factor Ultrabithorax is strongly influenced by various disordered regions that flank the structured homeodomain. Interactions with another, Extradenticle homeodomain are mediated by a short motif (shown by bold line) located in a clamp-like region (PDB code 1b8i). (D) Random complex: the cyclin-dependent kinase inhibitor Sic1 has nine phosphorylation sites that interchange upon contacting Cdc4. Contacts with two of them, T45 and S76, are shown by orange and magenta respectively. The phosphorylation sites are represented by spheres (as a courtesy of Dr Tanja Mittag). | ||
Here I review molecular mechanisms, which govern the formation of fuzzy complexes (Table 1). I analyze how dynamic regions contribute to fine-tuning of binding affinity and how it can be exploited for regulation. This provides a framework how ID proteins enable complex signaling pathways.
| Model | IDP | Partner | Category | Ref. |
|---|---|---|---|---|
| MeCP2: methylated CpG binding protein 2; TDG: thymine DNA glycosylase; ApLLP: aplysia LAPS18-like protein; SSB: single stranded DNA binding protein; myelin: myelin basic protein; p27: cell cycle kinase inhibitor p27; UmuD2: product of the umuD gene; PC4: human positive cofactor 4; FACT: facilitates chromatin transcription; HMGB1: high mobility group protein B1; Ubx: homeotic protein ultrabithorax; DSS1: small acidic Brh2-interacting protein; Brh2: BRCA2 analogue; NKX3.1: prostate tumor suppressor belonging to the NK_2 family; PPAR-γ: peroxisome proliferators activated receptor; UvrD: UvRD helicase; WH2: WASP-homology 2 domain (e.g. in Spire and Cordon-bleu); L7, L12: ribosomal proteins; UPF1: regulator of nonsense mediated decay; RPA: human replication protein A; KorB: transcriptional repressor. | ||||
| Conformational selection | ||||
| Max | DNA | F | 89 | |
| MeCP2 | DNA | F | 28 | |
| TDG | DNA | F | 30 | |
| Neurogenin | DNA | F | 90 | |
| ApLLP | DNA | F | 91 | |
| Flexibility modulation | ||||
| Ets-1 | DNA | F | 36 | |
| SSB | DNA | F | 92 | |
| Competitive binding | ||||
| RNase 1 | RNase inhibitor | P | 93 | |
| Myelin | Actin | P | 49 | |
| p27Kip1 | Cdk–cyclin | F | 38 | |
| β-Catenin | APC | F | 94 | |
| Sic1 | Cdc4 | R | 61 | |
| UmuD2 | UmuD2 | R | 95 | |
| Ebola virus nucleoprotein | VP35 and VP24 | F | 96 | |
| PC4 | DNA | F | 52 | |
| FACT | DNA | F | 60 | |
| HMGB1 | DNA | F | 97 | |
| Ubx | DNA | F, C | 40 | |
| DSS1/Brh2 | DNA | F | 98 | |
| NKX3.1 | DNA | F | 99 | |
| PPAR-γ | DNA | F | 100 | |
| UvrD | DNA | F | 101 | |
| β-Telomere | DNA | F | 102 | |
| Tethering | ||||
| WH2 | Actin | P | 103 | |
| L7 | L12 | C | 104 | |
| UPF1 | UPF2 | C | 42 | |
| RPA | DNA | C | 46 | |
| KorB | DNA | C | 105 | |
Box 1 Structural categories of fuzzy complexesIntrinsic disorder in fuzzy complexes, similarly to that in the free form comprises many different structural states along a wide spectrum, ranging from local to global disorder, from compact to extended shapes. Structural ambiguity could be static, where individual conformations could be resolved or dynamic, with inter-conversion among many conformations.13 Along these lines, fuzziness was classified into four categories.Polymorphic model (Fig. 1A)The bound IDP adopts unrelated conformations upon interacting with the same partner. The classical example is T cell factor 4 (Tcf4), where the acidic middle segment establishes alternative salt bridges with β-catenin, resulting in structural variability.84 Mutating any of the possible contacts impairs binding.Clamp model (Fig. 1B)Linkers connecting two globular binding regions preserve their conformational freedom in the bound form and do not establish direct interactions with the partner. Binding of scaffolding protein sterile 5 (Ste5p) to fusion 3 (Fus3p) protein,85 for example, can only be achieved in the presence of such a linker.Flanking model (Fig. 1C)ID segments often contact their partners via short motifs, while regions outside the binding context do not establish permanent contacts. These conformationally heterogeneous segments can increase binding affinity, as in the case of the kinase-inducible domain (KID) of cAMP-response element binding protein (CREB) upon interacting with CREB-binding protein (CBP).86Random model (Fig. 1D)As an extreme case of fuzziness, no regular structure is adopted upon contacting the partner. Furthermore, the NMR spectra are almost identical with those in the unbound form. A pertinent example is the oligomerization of T-cell receptor ζ-chains, through their disordered cytoplasmic domains.87Various IDPs, like TADs,88 the histone tails and yeast prions can bind to a partner without strict sequence requirements, i.e. their sequence can be scrambled without impacting function. Although direct structural evidence is lacking, they likely belong to fuzzy complexes. |
Dynamic recognition mechanisms in fuzzy complexes
IDPs interact with their partners via short motifs, which are termed by a variety of names (Table 2). These recognition sites can be divided into two major groups, depending on whether formation of a given structural element is required for binding. Preformed structural elements (PSE),16 pre-structured motifs (PreSMos)17 and molecular recognition features (MoRFs)18,19 transiently sample those conformations, which correspond to their bound state. Primary contact sites (PCS)20 and linear motifs (LMs)21 may not have residual structures, they mostly consists of hydrophobic residues embedded in a disordered environment. Binding sites of IDPs can be predicted using simple statistical potentials.22 Functioning all of these short binding sites on the one hand is intertwined with the plasticity of their environments,23 conformational equilibrium of which can be modulated to provide optimal fit with the partner. On the other hand interactions mediated by short motifs are usually weak24 and impart only a few constraints on the surrounding region. Consequently, the region embedding the ID binding site can retain most of its flexibility even in the bound form. The ID regions can be located almost in any place of the protein: flanking (flanking model) or in between folded regions (clamp model), or even spread around the protein at spatially and/or temporary varying locations (random model)13 (Box 1, Fig. 1).| Name | Abbreviation | Definition | Example IDP | Partner | Ref. |
|---|---|---|---|---|---|
| Preformed structural element | PSE | Transient secondary structure element that presages its bound state | p27Kip1 (37–59) | Cdk–cyclin | 16, 106 |
| Prestructured motif | PreSMo | Secondary structure motif, similar to the bound state | p53 TAD (18–26) | Mdm2 | 17, 107 |
| Molecular recognition element | α-MoRE | Folds into an α-helix upon binding | 4EBP1 (51–64) | eIF4E | 18, 108 |
| Molecular recognition feature | MoRF | Folds into a given secondary structure element upon binding | Med8 (193–210) | Med18/Med20 | 19, 109 |
| Primary contact site | PCS | Exposed site, which makes the first contacts with the partner | Calpastatin (135–146) | Calpain | 20, 110 |
| Linear motif | LM | Short, low complexity motif, which is responsible for specificity | Tau (547–553) | SH3 domain | 21, 23 |
| Anchor binding site | — | Folds upon binding | WASP (232–308) | Cdc42 | 22, 111 |
The most recent examples,14,15 where the ID segment not or only partly folds upon binding yet impacts the function of the complex, are listed in Table 1. These complexes include both protein–protein and protein–DNA interactions (examples discussed in the original ref. 13 were excluded). Structural and biochemical analysis of these complexes revealed four mechanisms how an ID segment can modulate specificity/affinity of the fuzzy complex without being actually involved in the binding interface. I will not discuss those examples, when the ID segment directly interacts with the partner, while it preserves its conformational ambiguity.
Conformational selection
ID segments can transiently sample conformations, which correspond to their bound forms. Shifting conformational equilibrium towards these residual structures thereby improves binding affinity. This mechanism is referred to as conformational selection (Fig. 2A).25 ID regions within the same protein can modulate the propensity of the binding-competent structure, which is not necessarily a regular motif. In all examples of this category so far, the regulating ID region flanks the binding segment.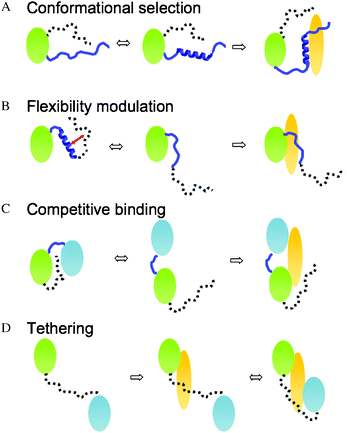 | ||
| Fig. 2 Four mechanisms how ID segments modulate binding via transient interactions. (A) Conformational selection: the ID segment shifts conformational equilibrium of the interface towards a binding-competent form. (B) Flexibility modulation: the ID segment frustrates the interaction network, which includes the binding region. This modulates the flexibility of the interface and in turn, the binding affinity. (C) Competitive binding: the ID segment interacts with the binding region and masks the contacting residues. (D) Tethering: the ID segments anchor the second binding region (e.g. weak-affinity region) to the partner. | ||
Methylated CpG binding protein 2 (MeCP2) deciphers epigenetic information by selectively recognizing DNA methylation patterns.26 Mutations of the protein lead to Rett syndrome, a severe neurodevelopmental disorder, which originates from changes in chromatin structure and gene repression. MeCP2 comprises various DNA binding domains, out of which the methylated DNA binding domain (MBD) recognizes the methylated CpG sequences.27 The propensity of regular secondary structures is 35% in the free MeCP2, which increases only by 7% upon interacting with DNA.28 This indicates that although some secondary structure elements are distinguished in binding, MeCP2 remains primarily disordered in the complex. The secondary structure content of the disordered N-terminal domain (NTD), for example, exhibits almost no change in the effect of DNA, yet it impacts affinity of the structured MBD by 10-fold.28 The NTD itself does not harbor a DNA binding site; but via inter-domain interactions it selects MBD conformations favorable for DNA binding. An even more profound effect is seen in the intervening domain and transcriptional regulatory domain, where the disordered C-terminal region helps to populate binding-competent conformations in the ensemble resulting in 30% improvement in DNA binding affinity. In line with their proposed role in mediating inter-domain contacts, a large number of short recognition sites were identified by bioinformatics analysis in these ID regions.28
A conformational transition is also coupled to the activity of thymine DNA glycosylase (TDG), which repairs GT and GU mismatches in the base excision repair (BER) pathway. TDG exist in two forms: the “closed” form, when the catalytic domain is involved in intra-molecular interactions, and an “open” form, which lacks these contacts.29 The turnover of this enzyme is regulated by the disordered N- and C-terminal domains (NTD and CTD), the removal of which increases the turnover of GT and GU activities by 50% and 100%, respectively.30 A 60-residue segment of the NTD, also termed as a regulatory domain (RD), adopts a stable secondary structure upon contacting the catalytic domain. The first 50 residues of the NTD and the whole CTD, however, remain disordered in the complex. The NTD was proposed to facilitate the transition towards the “closed” form of TDG, which is the DNA-binding competent conformation. Due to alternative recognition modes, this arrangement is more favorable for GT than for GU repair. Hence the disordered NTD regulates these two activities by shifting population between the “open” and “closed” conformations. In addition, both the N- and C-terminal residues compete for DNA contact sites hampering interactions between TDG and DNA.
Flexibility modulation
Long-range changes by distant ID regions in dynamic, but not structural properties of the protein can also influence interactions with the partner. Motions on the μs–ms timescale can modulate the flexibility of the residues at the interface thus altering the conformational entropy of binding (Fig. 2B). In addition to IDPs, this flexibility modulation mechanism takes place in various flexible, folded proteins, such as catabolic activator protein (CAP)31,32 or lactose repressor (Lac).33In response to Ca2+ signaling, Ets-1 transcription factor reduces DNA binding and transcription of Ets-1 driven reporters.34 The drop in binding affinity is caused by a serine and arginine rich region (SRR), which does not fold either in the absence or presence of the DNA. The SRR segment undergoes gradual phosphorylation upon Ca2+ release, but no secondary structures are induced.35 Phosphorylation, on the other hand, interferes with the intra-molecular interactions of the SRR region. These influence the contacts of SRR with the inhibitory module and the binding interface.36 By various measures, the hydrophobic network formed by the H3 recognition helices and HI-1 helix inhibitory module becomes more rigid with increasing phosphorylation of the SRR. The decreased flexibility prevents unfolding of the HI-1 helix, which is pre-requisite for binding. Thus the SRR, as a distant ID region in Ets-1, perturbs the dynamics of the protein–DNA interface and thereby attenuates binding.
Competitive binding
Fluctuations of the IDP chains can sterically block the access to the binding interface. Dynamic or static alternative interaction patterns may enable/disable interactions with other partners. Fuzzy complexes of all structural categories can function via the competitive binding mechanism (Fig. 2C).The p27Kip1 regulates eukaryotic cell division by interacting with cyclin-dependent kinases. In the Cdk2–cyclin complex, the p27 kinase inhibitory domain (KID) blocks the active site of the kinase.37 The dynamics of the interacting 310 helix segment, however, allows the exclusion of Y88 from the active site, which becomes available for phosphorylation by a nontyrosine-receptor kinase (Fig. 4).38 Due to the fuzziness of the C-terminal region, this segment flaps back onto the active site of Cdk2, and gets phosphorylated at T187 via a unimolecular mechanism. This phosphorylation event facilitates interactions with SCF/Skp2 ubiquitin ligase. Ubiquitination at the C-terminal region induces the degradation of p27Kip1 and causes the progression of the cell cycle.39 Hence the regulation by p27 is linked to two competitive mechanisms: (i) between Y88 and T187 to bind the active site of Cdk2, and (ii) between Cdk2/cyclin and SCF/Skp2 to bind T187. The latter is controlled by phosphorylation. Thus fuzziness in p27Kip1 underlies two opposite activities of the same protein.
Hox transcription factors control animal development through region specific differentiation. DNA recognition is carried out by the structured homeodomain (HD), which has high affinity, yet little selectivity in vivo.40,41 In the transcription factor Ultrabithorax (Ubx) multiple ID regions control DNA interactions (Fig. 1C). The inhibitory I1 and I2 regions reduce affinity of Ubx by 2-fold, and by 40-fold, respectively,40 whereas the R region improves binding in a length-dependent manner. pH-dependence of DNA binding indicates that I1 directly contacts ionizable residues of the HD binding interface and thus competes with DNA. I2 also acts via competitive mechanisms. On the one hand, I2 sterically blocks DNA contact sites of HD and on the other hand it competes with R for the same transient interactions.41 Such competitive interplay between I2 and R fine-tunes DNA binding affinity.
Tethering
In contrast to competing with residues involved in binding interfaces, ID regions can also improve binding affinity by enhancing cooperativity between globular binding domains.The complex of nonsense mediated decay factors UPF1 and UPF2 initiates mRNA degradation. UPF1 and UPF2 interact in a bipartite manner. The C terminal ID region of UPF2 adopts two regular secondary structures upon binding, which are connected by a linker without a rendered structure (poor electron density).42 This conforms to a clamp-like fuzzy model (Fig. 1B). The isolated α-helical segment of UPF2 has weaker affinity than the β-hairpin region, the cooperativity between the two segments, however, improves binding by 80-fold.42 Tethering by the linker region is not only beneficial for affinity, but also contributes to organization of the complex on a larger scale (Fig. 2D).43
The human replication protein A (RPA) is a single-stranded DNA binding protein, which participates in various DNA metabolic pathways including nucleotide excision repair and combinatorial repair.44 The 70 kDa subunit of human RPA (hRPA70) uses multiple domains for DNA interactions, which differ considerably in affinity. The weak and high affinity DNA binding domains (DBDs) are connected by a 78 amino acid long intrinsically unstructured linker domain (IULD).45 When RPA interacts with DNA, the first contacts are established with the high-affinity DNA binding domains. The weak affinity domain (DBD F) is anchored to DNA via the disordered IULD. This increases the local concentration of DBD F near the DNA and thus improves binding affinity. In accord, diminishing transient contacts of the IULD with DNA by mutations results in a 3-fold reduction in binding.46
Regulatory pathways enabled by fuzziness
Why fuzziness is beneficial for protein complexes? Dynamic/disordered regions impart versatility on the complex. This versatility stems from short functional motifs, which are embedded in conformationally heterogeneous regions in the bound state. All these short functional sites (i) modulate the number or type of partners in the complex, (ii) enable ultra-sensitive regulation of the complex or (iii) serve as a basis of specialization (e.g. tissue, context etc.) via mediating further interactions, post-translational modifications or alternative splicing (Fig. 3). These activities are not independent; post-translational modifications for example can also result in alternative interaction patterns.39,47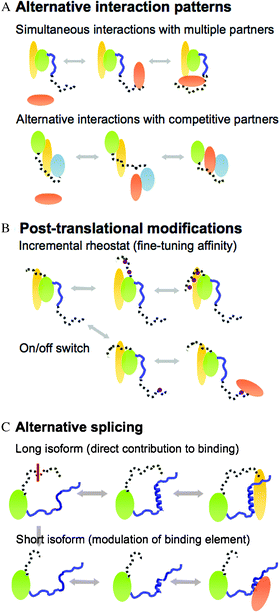 | ||
| Fig. 3 Three basic regulatory schemes enabled by fuzziness. (A) Alternative interaction patterns: simultaneous interactions with multiple partners (upper) or alternative contacts with different partners (lower). (B) Regulation by post-translational modifications, which fine-tune the affinity of the complex (upper) or work as an on/off switch (lower) (covalent attachments are shown by purple circles). (C) Alternative splicing, which changes the length of the fuzzy region. The long isoform directly contributes to binding (upper), whereas the short isoform modulates the conformational equilibrium of the binding region (lower). | ||
Alternative or simultaneous protein–protein interactions
Due to the plasticity of the ID region, the same motif can adapt to different partners or alternative patterns of the recognition motifs can mediate interactions with different partners (Fig. 3A). The myelin basic protein (MBP) is part of the mammalian central nervous system that plays a role in maintaining the stability and integrity of the myelin sheath. Myelin signaling is achieved via direct interactions with a variety of molecules, such as calmodulin, actin and tubulin. The proline-rich segment of the membrane bound MBP interacts with diverse SH3 domain-containing ligands (e.g. Yes1, PSD95, cortactin, PexD, Abl, Fyn, c-Src, Itk).48 The SH3 domains are typical components of intracellular signaling proteins, including nonreceptor tyrosine kinases, out of which Fyn also influences MBP expression. Besides the direct contacts with the proline residues, formation of all these complexes is facilitated by long-range electrostatic interactions between the ID regions of MBP outside the binding context.49 These interactions in combination with post-translational modifications and alternative splicing contribute to multifunctionality of MBP, and enable to integrate diverse signals from a variety of signaling pathways.50The human positive cofactor 4 (PC4) is a DNA-binding protein that is involved in transcriptional regulation by recruiting general transcription factors to the preinitiation complex. The activity of PC4 is strongly influenced by two ID segments: a serine- and acidic rich (SEAC) and a lysine-rich region, both located at the N-terminal domain (NTD).51 Although the NTD itself does not have a measurable affinity for DNA, it decreases the binding affinity of the C-terminal region of PC4 for ssDNA and reduces DNA unwinding activity of PC4.52 This is due to transient intra-molecular interactions between the NTD and the structured C-terminal domain (CTD), which compete with DNA binding. Phosphorylation of the lysine-rich region of the NTD strengthens these interactions, hence reduces binding affinity.53 PC4 also interacts with various trans-activator domains, for example, that of the herpes simplex virion protein 16 (VP16). Contacts with VP16 are also mediated by the disordered NTD, mostly via electrostatic attraction.52 The opposite effects of the NTD on the activator and DNA binding of PC4 indicate an overlap with the transient intra-molecular contacts of this region with CTD. Thus binding of an external partner interferes with the regulatory potential of this ID region.
Post-translational modifications, ultra-sensitivity
The activities of almost all fuzzy complexes (Table 1) are modulated by post-translational modifications.54 These covalent attachments, and phosphorylation in particular, can either play a role of an on/off switch between two states55 or gradually modulate affinity of the complex (Fig. 3B). In the first case phosphorylation beyond a given limit triggers the binding or the dissociation of a partner. Alternatively, multiple phosphorylation can serve as an ‘incremental rheostat’, by graded enhancement of binding via electrostatic interactions.36,56 Alternative post-translational modification patterns could serve as a “barcode” for different partners. This carries epigenetic information in the case of the N-terminal tail of H4 core histone protein.57,58Facilitates chromatin transcription (FACT) is involved in RNAP II-mediated transcription by displacing histone H2A–H2B dimers from nucleosomes. FACT interacts with DNA via a structured high-mobility group (HMG) domain, which is flanked by two charged ID regions of opposite character.59 The acidic ID region competes with DNA via intra-molecular interactions with both the HMG domain and the basic ID segment. Multiple phosphorylation of the acidic ID segment increases its negative charge and thus strengthens the interactions with the basic regions.60 This masks the nucleotide-binding elements, and results in inhibition of DNA binding. Phosphorylation, however, does not induce folding of the ID regions.
The complex of the cyclin-dependent kinase (CDK) inhibitor Sic1 and the SCF ubiquitin ligase subunit Cdc4 is formed in a phosphorylation dependent manner. Sic1 is a multivalent ligand comprising 9 phosphorylation sites, which target only a single binding site on Cdc4. Nevertheless, multiple phosphorylation (at least 6) is required for binding and subsequent degradation of Sic1.55 Phosphorylation causes only transient, local ordering around the binding motifs, while the whole complex remains dynamic.61 This enables an interchange between phosphorylation sites, which are in dynamic equilibrium with each other (Fig. 1D). The degree of phosphorylation fine-tunes the strength of interaction in the complex via long-range electrostatic interactions.62 Hence multiple phosphorylation ensue ultra-sensitivity of the Sic1–Cdc4 binding and ultimately, the cell cycle.
Alternative splicing, specialization
Protein activity can vary in a context-dependent manner. The same protein encounters different binding partners in different environments (cell, tissue) or the binding affinity of the same complex can depend on the milieu. Alternative splicing provides an elegant solution to these problems by altering the length of the ID regions and the embedded binding/post-translational modification sites in different isoforms63 (Fig. 3C).In MeCP2, the disordered NTD modulates the binding affinity for methylated CpG sequences (see above). Alternative translation start sites in murine MeCP2 increase the length of the NTD and thus perturb transient interactions with the structured MBD, which is responsible for methylated CpG recognition. Hence, the different isoforms lead to different gene expression patterns in mouse brain.64
Different splicing isoforms of the transcription factor Ubx are produced in a stage- and tissue-specific manner. Ubx contains three microexons. These are located next to the YPWM motif of the I1 inhibitory ID region, which mediates communication of the Extradenticle (Exd) cofactor65 (Fig. 1C). This interaction with Exd relieves the repression of DNA binding by I1.41 Removal of the microexons (alone or in combination) affects Ubx DNA affinity and selectivity41 both in vitro and in vivo.66,67 Based on these complex expression patterns, Ubx DNA binding affinity varies over a wide range depending on the cellular context.
Dynamic allostery in fuzzy complexes
Intrinsically disordered regions in the above listed regulatory pathways establish only transient contacts with the partner, thus could be considered as allosteric regulators. In the classical models of allostery, however, the dynamic equilibrium amongst these structures is shifted by selecting a conformer from the ensemble, which is engaged in interactions with a partner.25,68 Alternatively, the final, bound conformation is resulted by ‘induced fit’ on the effect of the partner.69 Protein dynamics in general has been considered as an important component in mediating signals.5,70 In dynamic allostery, the dynamic properties of the binding interface are modulated,32,71 by a flexible, but structured regulatory site. In contrast, the regulatory sites in fuzzy complexes preserve their conformational heterogeneity in the bound state54 (Fig. 3). This presents a novel case of allostery, which is associated with structural disorder of proteins.Here I propose to consider the disordered regulatory sites as an extension of the dynamic allostery. The new model describes the protein by a conformational ensemble in both the unbound and bound states (Fig. 4). Transitions between these populations are induced by the incoming signals and can be realized via multiple pathways. Structural plasticity of the bound state implies that disordered/dynamic segments can be subjected to further modifications, which in turn modifies the energy landscape of the whole protein. In other words, inter-molecular interactions or post-translational modifications involving the disordered regions work as a dynamic relay, affecting population or flexibility of different conformers at the binding interface.
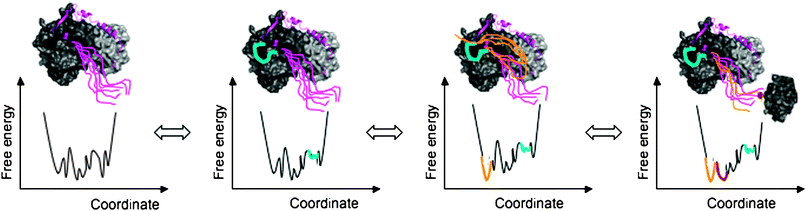 | ||
| Fig. 4 Allosteric regulation of fuzzy complexes. The complex is described by a conformational ensemble in all states. The conformational ensemble is represented by a free energy diagram as a function of a collective coordinate. Different interactions established by the disordered regions modulate the population and flexibility of the different conformers and thereby their contacts with further partners. Perturbation of the conformational ensemble is shown in colors corresponding to the respective disordered region (by cyan, yellow and red). In the complex of p27Kip1 (magenta) with Cdk2/cyclin (shown by dark and light grey; PDB code: 1jsu), for example, the C-terminal tail of p27 remains disordered (first panel). The region around Y88 (cyan), which interacts with the active site of Cdk2, is also flexible and can be ejected from the Cdk2 surface (second panel). This allows the disordered C-terminal tail to flip back (yellow conformers) and get phosphorylated by Cdk2 (third panel). Phosphorylation of T187 of the C-terminal tail (phosphate is shown by red) triggers interactions with SCF/Skp2 ubiquitin ligase (gray, fragment from PDB code: 2ast) (fourth panel). | ||
Conclusions and perspectives
The idea that protein complexes contain a significant amount of operative disorder is in sharp contrast with the classical view of one sequence → one structure → one protein function. The connection between conformational diversity and functional promiscuity has already been recognized.72–74 According to the ‘new view’,75 the existence of a single protein in multiple, instead of a single conformation underlies various activities. Hence, preserving the conformational heterogeneity in complex form provides an additional layer for regulation, as further interactions, post-translational modifications and alternative splicing of these ID segments can modulate the conformational ensemble of the complex.The emerging one sequence → multiple structures → multiple functions model has two important implications. (i) It emphasizes that weak, transient interactions outside the binding context can be critical in determining specificity/affinity of the final complex. These interactions, however, cannot be deduced from the final, static structure of the complex. Hence for experimental techniques to be suitable to analyze dynamic segments and transient interactions should be applied.59,76–80 Even if high-resolution structural studies are not feasible, low-resolution techniques and/or biochemical approaches may provide valuable information. Alternatively, the activity of biochemically-manipulated sequences (targeted mutagenesis and/or deletion constructs) could be investigated, including dynamic regions that establish transient interactions with the binding interface. (ii) Fuzzy complexes open new approaches to modulate signaling pathways. Small molecule inhibitors, for example, can specifically bind to ID regions in complexes. The dimerization of Max transcription factor with c-Myc oncogene can be inhibited, for example, by small organic compounds.81 Due to the lack of dimerization, c-Myc also fails to inhibit DNA binding and elicit transcriptional response. This suggests that fuzzy segments can be of potential pharmaceutical interest, and they can be targeted specifically without perturbing the structured part of the protein.
Based on the high propensity of ID regions in eukaryotic organisms,82,83 fuzzy complexes are also likely to be abundant. Fuzziness imparts versatility and dynamism on protein interaction networks, which is exploited in regulatory pathways. Hence the experimental and computational analysis of fuzzy complexes can significantly contribute to our understanding of cellular processes at the molecular level and can provide new opportunities to interfere with pathological mechanisms.
Acknowledgements
M.F. was supported by EMBO ASTF-312-2011 fellowship.References
- D. D. Boehr, R. Nussinov and P. E. Wright, The role of dynamic conformational ensembles in biomolecular recognition, Nat. Chem. Biol., 2009, 5(11), 789–796 CrossRef CAS.
- K. Henzler-Wildman and D. Kern, Dynamic personalities of proteins, Nature, 2007, 450(7172), 964–972 CrossRef CAS.
- K. A. Henzler-Wildman, M. Lei, V. Thai, S. J. Kerns, M. Karplus and D. Kern, A hierarchy of timescales in protein dynamics is linked to enzyme catalysis, Nature, 2007, 450(7171), 913–916 CrossRef CAS.
- K. A. Henzler-Wildman, V. Thai, M. Lei, M. Ott, M. Wolf-Watz, T. Fenn, E. Pozharski, M. A. Wilson, G. A. Petsko, M. Karplus, C. G. Hubner and D. Kern, Intrinsic motions along an enzymatic reaction trajectory, Nature, 2007, 450(7171), 838–844 CrossRef CAS.
- C. J. Tsai, A. Del Sol and R. Nussinov, Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms, Mol. BioSyst., 2009, 5(3), 207–216 RSC.
- A. del Sol, C. J. Tsai, B. Ma and R. Nussinov, The origin of allosteric functional modulation: multiple pre-existing pathways, Structure (London), 2009, 17(8), 1042–1050 CAS.
- F. Raimondi, M. Orozco and F. Fanelli, Deciphering the deformation modes associated with function retention and specialization in members of the Ras superfamily, Structure (London), 2010, 18, 402–414 CAS.
- P. Romero, Z. Obradovic, C. R. Kissinger, J. E. Villafranca, E. Garner, S. Guillot and A. K. Dunker, Thousands of proteins likely to have long disordered regions, Pac. Symp. Biocomput., 1998, 3, 437–448 Search PubMed.
- P. E. Wright and H. J. Dyson, Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm, J. Mol. Biol., 1999, 293(2), 321–331 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Intrinsically unstructured proteins and their functions, Nat. Rev. Mol. Cell Biol., 2005, 6(3), 197–208 CrossRef CAS.
- P. Tompa, The interplay between structure and function in intrinsically unstructured proteins, FEBS Lett., 2005, 579(15), 3346–3354 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Coupling of folding and binding for unstructured proteins, Curr. Opin. Struct. Biol., 2002, 12, 54–60 CrossRef CAS.
- P. Tompa and M. Fuxreiter, Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions, Trends Biochem. Sci., 2008, 33(1), 2–8 CrossRef CAS.
- M. Fuxreiter, I. Simon and S. Bondos, Dynamic protein–DNA recognition: beyond what can be seen, Trends Biochem. Sci., 2011, 36(8), 415–423 CrossRef CAS.
- M. Fuxreiter and P. Tompa, Fuzziness: Structural Disorder in Protein Complexes Austin, Landes BioScience/Springer, New York, 2011 Search PubMed.
- M. Fuxreiter, I. Simon, P. Friedrich and P. Tompa, Preformed structural elements feature in partner recognition by intrinsically unstructured proteins, J. Mol. Biol., 2004, 338(5), 1015–1026 CrossRef CAS.
- S.-H. Lee, D.-H. Kim, J. J. Han, E.-J. Cha, J.-E. Lim, Y.-J. Cho, C. Lee and K.-H. Han, Understanding Pre-Structured Motifs (PreSMos) in intrinsically unfolded proteins, Curr. Protein Pept. Lett., 2011 Search PubMed , in press.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Coupled folding and binding with alpha-helix-forming molecular recognition elements, Biochemistry, 2005, 44(37), 12454–12470 CrossRef CAS.
- A. Mohan, C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker and V. N. Uversky, Analysis of molecular recognition features (MoRFs), J. Mol. Biol., 2006, 362(5), 1043–1059 CrossRef CAS.
- V. Csizmok, M. Bokor, P. Banki, E. Klement, K. F. Medzihradszky, P. Friedrich, K. Tompa and P. Tompa, Primary contact sites in intrinsically unstructured proteins: the case of calpastatin and microtubule-associated protein 2, Biochemistry, 2005, 44(10), 3955–3964 CrossRef CAS.
- P. Puntervoll, R. Linding, C. Gemund, S. Chabanis-Davidson, M. Mattingsdal, S. Cameron, D. M. Martin, G. Ausiello, B. Brannetti, A. Costantini, F. Ferre, V. Maselli, A. Via, G. Cesareni, F. Diella, G. Superti-Furga, L. Wyrwicz, C. Ramu, C. McGuigan, R. Gudavalli, I. Letunic, P. Bork, L. Rychlewski, B. Kuster, M. Helmer-Citterich, W. N. Hunter, R. Aasland and T. J. Gibson, ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins, Nucleic Acids Res., 2003, 31(13), 3625–3630 CrossRef CAS.
- B. Meszaros, I. Simon and Z. Dosztanyi, Prediction of protein binding regions in disordered proteins, PLoS Comput. Biol., 2009, 5(5), e1000376 Search PubMed.
- M. Fuxreiter, P. Tompa and I. Simon, Local structural disorder imparts plasticity on linear motifs, Bioinformatics, 2007, 23(8), 950–956 CrossRef CAS.
- V. Neduva and R. B. Russell, Linear motifs: evolutionary interaction switches, FEBS Lett., 2005, 579(15), 3342–3345 CrossRef CAS.
- C. Tsai, B. Ma, Y. Y. Sham, S. Kumar and R. Nussinov, Structured disorder and conformational selection, Proteins: Struct., Funct., Genet., 2001, 44, 418–427 CrossRef CAS.
- J. C. Hansen, R. P. Ghosh and C. L. Woodcock, Binding of the Rett syndrome protein, MeCP2, to methylated and unmethylated DNA and chromatin, IUBMB Life, 2010, 62(10), 732–738 CrossRef CAS.
- V. H. Adams, S. J. McBryant, P. A. Wade, C. L. Woodcock and J. C. Hansen, Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2, J. Biol. Chem., 2007, 282(20), 15057–15064 CrossRef CAS.
- R. P. Ghosh, T. Nikitina, R. A. Horowitz-Scherer, L. M. Gierasch, V. N. Uversky, K. Hite, J. C. Hansen and C. L. Woodcock, Unique physical properties and interactions of the domains of methylated DNA binding protein 2, Biochemistry, 2010, 49(20), 4395–4410 CrossRef CAS.
- R. Steinacher and P. Schar, Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation, Curr. Biol., 2005, 15, 616–623 CrossRef CAS.
- C. Smet-Nocca, J. M. Wieruszeski, V. Chaar, A. Leroy and A. Benecke, The thymine-DNA glycosylase regulatory domain: residual structure and DNA binding, Biochemistry, 2008, 47(25), 6519–6530 CrossRef CAS.
- N. Popovych, S. R. Tzeng, M. Tonelli, R. H. Ebright and C. G. Kalodimos, Structural basis for cAMP-mediated allosteric control of the catabolite activator protein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(17), 6927–6932 CrossRef CAS.
- S. R. Tzeng and C. G. Kalodimos, Dynamic activation of an allosteric regulatory protein, Nature, 2009, 462(7271), 368–372 CrossRef CAS.
- C. G. Kalodimos, N. Biris, A. M. Bonvin, M. M. Levandoski, M. Guennuegues, R. Boelens and R. Kaptein, Structure and flexibility adaptation in nonspecific and specific protein–DNA complexes, Science, 2004, 305(5682), 386–389 CrossRef CAS.
- H. Liu and T. Grundström, Calcium regulation of GM-CSF by calmodulin-dependent kinase II phosphorylation of Ets1, Mol. Biol. Cell, 2002, 13, 4497–4507 CrossRef CAS.
- G. M. Lee, M. A. Pufall, C. A. Meeker, H. S. Kang, B. J. Graves and L. P. McIntosh, The affinity of Ets-1 for DNA is modulated by phosphorylation through transient interactions of an unstructured region, J. Mol. Biol., 2008, 382(4), 1014–1030 CrossRef CAS.
- M. A. Pufall, G. M. Lee, M. L. Nelson, H. S. Kang, A. Velyvis, L. E. Kay, L. P. McIntosh and B. J. Graves, Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region, Science, 2005, 309(5731), 142–145 CrossRef CAS.
- E. R. Lacy, I. Filippov, W. S. Lewis, S. Otieno, L. Xiao, S. Weiss, L. Hengst and R. W. Kriwacki, p27 binds cyclin–CDK complexes through a sequential mechanism involving binding-induced protein folding, Nat. Struct. Mol. Biol., 2004, 11(4), 358–364 CAS.
- C. A. Galea, A. Nourse, Y. Wang, S. G. Sivakolundu, W. T. Heller and R. W. Kriwacki, Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1, J. Mol. Biol., 2008, 376(3), 827–838 CrossRef CAS.
- C. A. Galea, Y. Wang, S. G. Sivakolundu and R. W. Kriwacki, Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits, Biochemistry, 2008, 47(29), 7598–7609 CrossRef CAS.
- Y. Liu, K. S. Matthews and S. E. Bondos, Multiple intrinsically disordered sequences alter DNA binding by the homeodomain of the Drosophila hox protein ultrabithorax, J. Biol. Chem., 2008, 283(30), 20874–20887 CrossRef CAS.
- Y. Liu, K. S. Matthews and S. E. Bondos, Internal regulatory interactions determine DNA binding specificity by a Hox transcription factor, J. Mol. Biol., 2009, 390(4), 760–774 CrossRef CAS.
- M. Clerici, A. Mourao, I. Gutsche, N. H. Gehring, M. W. Hentze, A. Kulozik, J. Kadlec, M. Sattler and S. Cusack, Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2, EMBO J., 2009, 28(15), 2293–2306 CrossRef CAS.
- P. V. Ivanov, N. H. Gehring, J. B. Kunz, M. W. Hentze and A. E. Kulozik, Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways, EMBO J., 2008, 27, 736–747 CrossRef CAS.
- M. S. Wold, Replication protein A: a heterotrimeric single-stranded DNA-binding protein required for eukaryotic DNA metabolism, Annu. Rev. Biochem., 1997, 66, 61–92 CrossRef CAS.
- K. E. Olson, P. Narayanaswami, P. D. Vise, D. F. Lowry, M. S. Wold and G. W. Daughdrill, Secondary structure and dynamics of an intrinsically unstructured linker domain, J. Biomol. Struct. Dyn., 2005, 23(2), 113–124 CAS.
- P. D. Vise, B. Baral, A. J. Latos and G. W. Daughdrill, NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain, Nucleic Acids Res., 2005, 33(7), 2061–2077 CrossRef CAS.
- Y. Wang, J. C. Fisher, R. Mathew, L. Ou, S. Otieno, J. Sublet, L. Xiao, J. Chen, M. F. Roussel and R. W. Kriwacki, Intrinsic disorder mediates the diverse regulatory functions of the Cdk inhibitor p21, Nat. Chem. Biol., 2011, 7, 214–221 CrossRef CAS.
- E. Polverini, G. Rangaraj, D. S. Libich, J. M. Boggs and G. Harauz, Binding of the proline-rich segment of myelin basic protein to SH3 domains: spectroscopic, microarray, and modeling studies of ligand conformation and effects of post-translational modifications, Biochemistry, 2008, 47(1), 267–282 CrossRef CAS.
- M. A. Ahmed, V. V. Bamm, L. Shi, M. Steiner-Mosonyi, J. F. Dawson, L. Brown, G. Harauz and V. Ladizhansky, Induced secondary structure and polymorphism in an intrinsically disordered structural linker of the CNS: solid-state NMR and FTIR spectroscopy of myelin basic protein bound to actin, Biophys. J., 2009, 96(1), 180–191 CrossRef CAS.
- C. M. D. Hill, D. S. Libich and G. Harauz, Assembly of tubulin by classic myelin basic protein isoforms and regulation by post-translational modification, Biochemistry, 2005, 44, 16672–16683 CrossRef CAS.
- B. R. Kumar, V. Swaminathan, S. Banerjee and T. K. Kundu, p300-mediated acetylation of human transcriptional coactivator PC4 is inhibited by phosphorylation, J. Biol. Chem., 2001, 276(20), 16804–16809 CrossRef CAS.
- H. R. Jonker, R. W. Wechselberger, R. Boelens, R. Kaptein and G. E. Folkers, The intrinsically unstructured domain of PC4 modulates the activity of the structured core through inter- and intramolecular interactions, Biochemistry, 2006, 45(15), 5067–5081 CrossRef CAS.
- H. R. Jonker, R. W. Wechselberger, M. Pinkse, R. Kaptein and G. E. Folkers, Gradual phosphorylation regulates PC4 coactivator function, FEBS J., 2006, 273(7), 1430–1444 CrossRef CAS.
- M. Fuxreiter, P. Tompa, I. Simon, V. N. Uversky, J. C. Hansen and F. J. Asturias, Malleable machines take shape in eukaryotic transcriptional regulation, Nat. Chem. Biol., 2008, 4(12), 728–737 CrossRef CAS.
- P. Nash, X. Tang, S. Orlicky, Q. Chen, F. B. Gertler, M. D. F. Sicheri, T. Pawson and M. Tyers, Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication, Nature, 2001, 414(6863), 514–521 CrossRef CAS.
- C. W. Lee, J. C. Ferreon, A. C. Ferreon, M. Arai and P. E. Wright, Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(45), 19290–19295 CrossRef CAS.
- T. Jenuwein and C. D. Allis, Translating the histone code, Science, 2001, 293(5532), 1074–1080 CrossRef CAS.
- M. Shogren-Knaak, H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie and C. L. Peterson, Histone H4-K16 acetylation controls chromatin structure and protein interactions, Science, 2006, 311(5762), 844–847 CrossRef CAS.
- A. Miyagi, Y. Tsunaka, T. Uchihashi, K. Mayanagi, S. Hirose, K. Morikawa and T. Ando, Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy, ChemPhysChem, 2008, 9(13), 1859–1866 CrossRef CAS.
- Y. Tsunaka, J. Toga, H. Yamaguchi, S. Tate, S. Hirose and K. Morikawa, Phosphorylated intrinsically disordered region of FACT masks its nucleosomal DNA binding elements, J. Biol. Chem., 2009, 284(36), 24610–24621 CrossRef CAS.
- T. Mittag, S. Orlicky, W. Y. Choy, X. Tang, H. Lin, F. Sicheri, L. E. Kay, M. Tyers and J. D. Forman-Kay, Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(46), 17772–17777 CrossRef CAS.
- M. Borg, T. Mittag, T. Pawson, M. Tyers, J. D. Forman-Kay and H. S. Chan, Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(23), 9650–9655 CrossRef CAS.
- S. E. Pursglove, M. Fladvad, M. Bellanda, A. Moshref, M. Henriksson, J. Carey and M. Sunnerhagen, Biophysical properties of regions flanking the bHLH-Zip motif in the p22 Max protein, Biochem. Biophys. Res. Commun., 2004, 323(3), 750–759 CrossRef CAS.
- J. M. Dragich, Y. H. Kim, A. P. Arnold and N. C. Schanen, Differential distribution of the MeCP2 splice variants in the postnatal mouse brain, J. Comp. Neurol., 2007, 501(4), 526–542 CrossRef.
- J. M. Passner, H. D. Ryoo, L. Shen, R. S. Mann and A. K. Aggarwal, Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex, Nature, 1999, 397(6721), 714–719 CrossRef CAS.
- H. C. Reed, T. Hoare, S. Thomsen, T. A. Weaver, R. A. White, M. Akam and C. R. Alonso, Alternative splicing modulates Ubx protein function in Drosophila melanogaster, Genetics, 2010, 184(3), 745–758 CrossRef CAS.
- E. Tour, C. T. Hittinger and W. McGinnis, Evolutionarily conserved domains required for activation and repression functions of the Drosophila Hox protein Ultrabithorax, Development (Cambridge, UK), 2005, 132(23), 5271–5281 CAS.
- J. Monod, J. Wyman and J. P. Changeux, On the Nature of Allosteric Transitions: A Plausible Model, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS.
- D. E. Koshland Jr., G. Nemethy and D. Filmer, Comparison of experimental binding data and theoretical models in proteins containing subunits, Biochemistry, 1966, 5(1), 365–385 CrossRef CAS.
- R. G. Smock and L. M. Gierasch, Sending signals dynamically, Science, 2009, 324(5924), 198–203 CrossRef CAS.
- N. Popovych, S. Sun, R. H. Ebright and C. G. Kalodimos, Dynamically driven protein allostery, Nat. Struct. Mol. Biol., 2006, 13(9), 831–838 CAS.
- B. Ma, M. Shatsky, H. J. Wolfson and R. Nussinov, Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations, Protein Sci., 2009, 11, 184–197 CrossRef.
- S. D. Copley, Enzymes with extra talents: moonlighting functions and catalytic promiscuity, Curr. Opin. Chem. Biol., 2003, 7, 265–272 CrossRef CAS.
- L. C. James, P. Roversi and D. S. Tawfik, Antibody multispecificity mediated by conformational diversity, Science, 2003, 299(5611), 1362–1367 CrossRef CAS.
- L. C. James and D. S. Tawfik, Conformational diversity and protein evolution—a 60-year-old hypothesis revisited, Trends Biochem. Sci., 2003, 28(7), 361–368 CrossRef CAS.
- J. A. Marsh and J. D. Forman-Kay, Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints, J. Mol. Biol., 2009, 391(2), 359–374 CrossRef CAS.
- M. R. Jensen, G. Communie, E. A. J. Ribeiro, N. Martinez, A. Desfosses, L. Salmon, L. Mollica, F. Gabel, M. Jamin, S. Longhi, R. W. Ruigrok and M. Blackledge, Intrinsic disorder in measles virus nucleocapsids, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(24), 9839–9844 CrossRef CAS.
- M. R. Jensen, G. Communie, E. A. Ribeiro Jr., N. Martinez, A. Desfosses, L. Salmon, L. Mollica, F. Gabel, M. Jamin, S. Longhi, R. W. Ruigrok and M. Blackledge, Intrinsic disorder in measles virus nucleocapsids, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(24), 9839–9844 CrossRef CAS.
- I. Bertini, A. Giachetti, C. Luchinat, G. Parigi, M. V. Petoukhov, R. Pierattelli, E. Ravera and D. I. Svergun, Conformational space of flexible biological macromolecules from average data, J. Am. Chem. Soc., 2010, 132(38), 13553–13558 CrossRef CAS.
- A. C. Ferreon, C. R. Moran, Y. Gambin and A. A. Deniz, Single-molecule fluorescence studies of intrinsically disordered proteins, Methods Enzymol., 2010, 472, 179–204 CAS.
- A. V. Follis, D. I. Hammoudeh, H. Wang, E. V. Prochownik and S. J. Metallo, Structural rationale for coupled binding and unfolding of the c-Myc oncoprotein by small molecules, Chem. Biol., 2008, 15, 1149–1155 CrossRef CAS.
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, Prediction and functional analysis of native disorder in proteins from the three kingdoms of life, J. Mol. Biol., 2004, 337(3), 635–645 CrossRef CAS.
- P. Tompa, Z. Dosztanyi and I. Simon, Prevalent structural disorder in E. coli and S. cerevisiae proteomes, J. Proteome Res., 2006, 5(8), 1996–2000 CrossRef CAS.
- T. A. Graham, D. M. Ferkey, F. Mao, D. Kimelman and W. Xu, Tcf4 can specifically recognize beta-catenin using alternative conformations, Nat. Struct. Biol., 2001, 8(12), 1048–1052 CrossRef CAS.
- R. P. Bhattacharyya, A. Remenyi, M. C. Good, C. J. Bashor, A. M. Falick and W. A. Lim, The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway, Science, 2006, 311(5762), 822–826 CrossRef CAS.
- T. Zor, B. M. Mayr, H. J. Dyson, M. R. Montminy and P. E. Wright, Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators, J. Biol. Chem., 2002, 277(44), 42241–42248 CrossRef CAS.
- A. Sigalov, D. Aivazian and L. Stern, Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif, Biochemistry, 2004, 43(7), 2049–2061 CrossRef CAS.
- P. B. Sigler, Transcriptional activation. Acid blobs and negative noodles, Nature, 1988, 333(6170), 210–212 CrossRef CAS.
- J. F. Naud, F. O. McDuff, S. Sauve, M. Montagne, B. A. Webb, S. P. Smith, B. Chabot and P. Lavigne, Structural and thermodynamical characterization of the complete p21 gene product of Max, Biochemistry, 2005, 44(38), 12746–12758 CrossRef CAS.
- D. Aguado-Llera, E. Goormaghtigh, N. de Geest, X. J. Quan, A. Prieto, B. A. Hassan, J. Gomez and J. L. Neira, The basic helix–loop–helix region of human neurogenin 1 is a monomeric natively unfolded protein which forms a “fuzzy” complex upon DNA binding, Biochemistry, 2010, 49(8), 1577–1589 CrossRef CAS.
- J. Liu and J. Song, A novel nucleolar transcriptional activator ApLLP for long-term memory formation is intrinsically unstructured but functionally active, Biochem. Biophys. Res. Commun., 2008, 366(2), 585–591 CrossRef CAS.
- S. N. Savvides, S. Raghunathan, K. Futterer, A. G. Kozlov, T. M. Lohman and G. Waksman, The C-terminal domain of full-length E. coli SSB is disordered even when bound to DNA, Protein Sci., 2004, 13(7), 1942–1947 CrossRef CAS.
- K. E. Kover, M. Bruix, J. Santoro, G. Batta, D. V. Laurents and M. Rico, The solution structure and dynamics of human pancreatic ribonuclease determined by NMR spectroscopy provide insight into its remarkable biological activities and inhibition, J. Mol. Biol., 2008, 379(5), 953–965 CrossRef CAS.
- N. C. Ha, T. Tonozuka, J. L. Stamos, H. J. Choi and W. I. Weis, Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation, Mol. Cell, 2004, 15(4), 511–521 CrossRef CAS.
- S. M. Simon, F. J. Sousa, R. Mohana-Borges and G. C. Walker, Regulation of Escherichia coli SOS mutagenesis by dimeric intrinsically disordered umuD gene products, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(4), 1152–1157 CrossRef CAS.
- W. Shi, Y. Huang, M. Sutton-Smith, B. Tissot, M. Panico, H. R. Morris, A. Dell, S. M. Haslam, J. Boyington, B. S. Graham, Z. Y. Yang and G. J. Nabel, A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions, J. Virol., 2008, 82(13), 6190–6199 CrossRef CAS.
- K. Stott, M. Watson, F. S. Howe, J. G. Grossmann and J. O. Thomas, Tail-mediated collapse of HMGB1 is dynamic and occurs via differential binding of the acidic tail to the A and B domains, J. Mol. Biol., 2010, 403(5), 706–722 CrossRef CAS.
- Q. Zhou, N. Mazloum, N. Mao, M. Kojic and W. K. Holloman, Dss1 regulates interaction of Brh2 with DNA, Biochemistry, 2009, 48(50), 11929–11938 CrossRef CAS.
- J. H. Ju, J. S. Maeng, D. Y. Lee, G. Piszczek, E. P. Gelmann and J. M. Gruschus, Interactions of the acidic domain and SRF interacting motifs with the NKX3.1 homeodomain, Biochemistry, 2009, 48(44), 10601–10607 CrossRef CAS.
- V. Chandra, P. Huang, Y. Hamuro, S. Raghuram, Y. Wang, T. P. Burris and F. Rastinejad, Structure of the intact PPAR-gamma-RXR-nuclear receptor complex on DNA, Nature, 2008, 456(7220), 350–356 CrossRef.
- L. Manelyte, C. P. Guy, R. M. Smith, M. S. Dillingham, P. McGlynn and N. J. Savery, The unstructured C-terminal extension of UvrD interacts with UvrB, but is dispensable for nucleotide excision repair, DNA Repair, 2009, 8(11), 1300–1310 CrossRef CAS.
- P. Buczek and M. P. Horvath, Structural reorganization and the cooperative binding of single-stranded telomere DNA in Sterkiella nova, J. Biol. Chem., 2006, 281(52), 40124–40134 CrossRef CAS.
- L. Renault, B. Bugyi and M. F. Carlier, Spire and Cordon-bleu: multifunctional regulators of actin dynamics, Trends Cell Biol., 2008, 18(10), 494–504 CrossRef CAS.
- M. G. Bubunenko, S. V. Chuikov and A. T. Gudkov, The length of the interdomain region of the L7/L12 protein is important for its function, FEBS Lett., 1992, 313(3), 232–234 CrossRef CAS.
- K. Rajasekar, S. T. Muntaha, J. R. Tame, S. Kommareddy, G. Morris, C. W. Wharton, C. M. Thomas, S. A. White, E. I. Hyde and D. J. Scott, Order and disorder in the domain organization of the plasmid partition protein KorB, J. Biol. Chem., 2010, 285(20), 15440–15449 CrossRef CAS.
- S. G. Sivakolundu, D. Bashford and R. W. Kriwacki, Disordered p27Kip1 exhibits intrinsic structure resembling the Cdk2/cyclin A-bound conformation, J. Mol. Biol., 2005, 353(5), 1118–1128 CrossRef CAS.
- S. W. Chi, S. H. Lee, D. H. Kim, M. J. Ahn, J. S. Kim, J. Y. Woo, T. Torizawa, M. Kainosho and K. H. Han, Structural details on mdm2-p53 interaction, J. Biol. Chem., 2005, 280(46), 38795–38802 CrossRef CAS.
- J. Marcotrigiano, A. C. Gingras, N. Sonenberg and S. K. Burley, Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G, Mol. Cell, 1999, 3(6), 707–716 CrossRef CAS.
- L. Lariviere, S. Geiger, S. Hoeppner, S. Rother, K. Strasser and P. Cramer, Structure and TBP binding of the Mediator head subcomplex Med8-Med18-Med20, Nat. Struct. Mol. Biol., 2006, 13(10), 895–901 CAS.
- T. Moldoveanu, K. Gehring and D. R. Green, Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterotrimetic calpains, Nature, 2008, 456, 404–408 CrossRef CAS.
- N. Abdul-Manan, B. Aghazadeh, G. A. Liu, A. Majumdar, O. Ouerfelli, K. A. Siminovitch and M. K. Rosen, Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott–Aldrich syndrome’ protein, Nature, 1999, 399(6734), 379–383 CrossRef CAS.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |