Overcoming the PEG-addiction: well-defined alternatives to PEG, from structure–property relationships to better defined therapeutics
Matthias
Barz†
*ab,
Robert
Luxenhofer†
*c,
Rudolf
Zentel
b and
María J.
Vicent
a
aCentro de Investigación Príncipe Felipe, Polymer Therapeutics Lab, Avda Autopista del Saler 16/3, 46012, Valencia, Spain. E-mail: mbarz@cipf.es; Fax: +34 963289701; Tel: +34 963289680
bInstitute of Organic Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55099, Mainz, Germany. E-mail: barz@uni-mainz.de; Fax: +49 6131 39 24778; Tel: +49 6131 39 25468
cProfessur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Zellescher Weg 19, 01069, Dresden, Germany. E-mail: robert.luxenhofer@chemie.tu-dresden.de; Fax: +49 351 463 37122; Tel: +49 351 463 36057
First published on 28th March 2011
Abstract
Synthetic methods in polymer chemistry have evolved tremendously during the last decade. Nowadays more and more attention is devoted to the application of those tools in the development of the next generation of nanomedicines. Nevertheless, poly(ethylene glycol) (PEG) remains the most frequently used polymer for biomedical applications. In this review, we try to summarize recent efforts and developments in controlled polymerisation techniques that may allow alternatives to PEG based systems and can be used to improve the properties of future polymer therapeutics.
 Matthias Barz | Dr Matthias Barz finished his PhD in 2009 working on the synthesis, radioactive labeling and functionalisation of biocompatible polymer architectures under the supervision of Prof. Rudolf Zentel. In 2010 he joined the group of Dr María Jesus Vicent at the CIPF in Valencia (Spain) as a postdoctoral research fellow. During his time in the polymer therapeutics lab of Maria J. Vicent he worked on the development of enzyme specific imaging probes based on poly(glutamic acid). After returning to Germany, he is currently working as a research fellow at the institute of organic chemistry at the Johannes Gutenberg-University Mainz. His research interests are the synthesis of functional polymers based on natural and non-natural building blocks, the development of selective functionalisation strategies as well as the polymer supported transport and directed intracellular translocation of bioactive agents. |
 Robert Luxenhofer | Dr Robert Luxenhofer is a research fellow of the King Abdullah University of Science and Technology (KAUST) at the Technische Universität Dresden. In 2007 he completed his PhD at the Technische Universität München in the field of polymer chemistry under the supervision of Rainer Jordan. After postdoctoral work on drug delivery and endocytosis of polymers with Alexander Kabanov at the University of Nebraska Medical Center (Omaha, USA), he moved to Technische Universität Dresden in 2009. His research interests include synthesis of tailored polymers for biomedical applications and interaction between polymers and mammalian cells. |
 Rudolf Zentel | Prof. Rudolf Zentel studied chemistry at the University of Mainz, where he finished his PhD thesis in 1983 jointly supervised by professors Ringsdorf and Strobl. During this time he got introduced to the concept of polymers for pharmaceutical applications. After a postdoctoral stay (Freiburg), Habilitation (Mainz 1989) and research stays at the “IBM Almaden Research Center” (San Jose, USA, 1989–1990) and in Düsseldorf (1990–1992) he got his first professorship 1992 in Mainz. After an intermezzo at the university of Wuppertal (chair for Material Science 1996–2000) he returned to the university of Mainz in 2000. Since 2006 he is the German speaker of the International Research Training Group “Self-organised Materials for Optoelectronics”, Mainz—Seoul. Central topics of his research are self-organizing systems (LC-phases or colloids), the interaction of matter with light and—recently again—well defined biocompatible polymers as nanocarriers for bioactive agents. For more details see: http://www.zentel.de/. |
 María J. Vicent | María J. Vicent is the Head of the Polymer Therapeutics Laboratory in the Chemical Biology Dept at Príncipe Felipe Research Center. After a PhD in material sciences between Castellón (Spain, Luis' Lab.) and U.C. Berkeley (USA, Frechet's Lab.), a Marie Curie funded postdoc in Prof. Duncan's Lab. (Cardiff, UK) and one-year research associate position through a Marie Curie Reintegration contract at CIPF (Valencia, Spain); she was appointed as principal investigator in June 2006 to build the first Polymer Therapeutics Lab. in Spain. She has published more than 50 papers and book's chapters and she's also a named inventor in 4 patents 2 of them already licensed. María was awarded with a Prize on Basic Sciences from ‘Fundación de las Artes y las Ciencias’ (‘IV Edition Idea Awards’ Valencia, Spain) in June 2008. She is the Spanish President of the Spanish-Portuguese Chapter of the Controlled Release Society and is chairing the Polymer Therapeutics Symposium: from Lab to Clinic, one of the most recognised conferences in the field. |
1. Introduction
Modern life as we know it would be simply impossible without polymers. Natural and synthetic polymers are essential not only in our day-to-day life but have also become increasingly important in biomedical applications. To our knowledge the first polymer–drug conjugate dates back already more than half a century1 and an early rationale for polymer conjugates for therapeutic applications was published several decades ago in a visionary work of H. Ringsdorf,2 while the terms polymer therapeutics3 and nanomedicine4 have come into use only recently.While nature is preparing and using defined multifunctional polymers, i.e.polypeptides, -saccharides and -nucleotides since the dawn of biotic times, humans have been consciously preparing polymers only for about a century and well defined polymers are still playing a minor role outside research laboratories. With the prominent exception of poly(ethylene glycol) (PEG) practically no defined polymer platform is used in biomedical applications. This fact has had probably two main reasons. First, the difficulty to prepare defined polymers and polymeric systems, in particular in large scale, and, secondly and somehow as a consequence from the first reason, the lack of knowledge of the effects of polymer architecture, size, charge or charge distribution etc.in vitro and in vivo, factors that can only be assessed when defined polymers are used. However, in certain applications defined polymers are indispensable. Whenever polymers, especially non-degradable ones, are used for in vivo application as polymer–drug conjugates,5 micelles,6,7 polymersomes,8,9nanoparticles10–12 or protein–polymer conjugates13–16 we need to be aware of their in vivo fate. Are they cleared from the organism or do they (or more likely a certain fraction) accumulate, perhaps in a specific organ? This knowledge is of major importance in order to avoid side effects and long term toxicity in future nanopharmaceuticals. For example, the renal exclusion limit as an example depends, among other factors, on the size of polymers in solution. In this respect, macromolecules having comparable sizes are mandatory to fine-tune the properties of the whole population.
Up to date, the most commonly used well-defined polymer in biomedical applications is PEG in various architectures, especially after Webster et al. claimed the safety of PEG for medical application.17 More recently however, several linear reports show that under certain conditions PEG and PEG-containing polymers can elicit significant complement activation,18,19 and rapid clearance can occur after repeated injections of PEGylated liposomes.20 Moreover, as a polyether, PEG is prone to undergo peroxidation which may affect bioactives, cells and tissues in various ways.21–23 Not only for these reasons alternatives to PEG in biomedical applications are investigated. A huge number of reviews on PEG and its use are available.24,25 PEGylated proteins are already in the market and PEG based micelles in the advanced clinical phases. From the point of view of a polymer chemist especially the PEG based block copolymer micelles first described by Hörpel et al. in 1985 have opened the field for micellar drug delivery systems.26 This research area was later on shaped by the outstanding work of the groups of Kataoka and Kabanov.7,27,28 The rise of PEG to the ‘gold standard’ of water-soluble biomaterial may have had several reasons. One major, if not the most important reason, was its commercial availability in sufficient quality and in a wide range of molar masses. The majority of labs investigating polymers for biomedical applications simply lacked the capacity to prepare defined polymers, let alone PEG, where safe handling of the monomer is not trivial. However, PEG is not without alternatives. In this review we attempt to give an overview on what we believe to be some of the most interesting substitutes.
In the last two decades, a multitude of new methods for the controlled polymerisation techniques has been established. Especially the controlled radical polymerisation methods atom transfer radical polymerisation (ATRP),29 reversible addition fragmentation chain transfer (RAFT) polymerisation30 and nitroxide mediated polymerisation (NMP)31 have pushed this field enormously, giving access to defined polyacrylates and polyacrylamides. In addition, the preparation of defined, synthetic polypeptides has made huge progress since the first reports by Deming and coworkers.32 Poly(2-oxazoline)s (POx) are another type of polymers that can be synthesised in a defined manner since the 1950's, and have shown some promise for biomedical applications in the 1990's,33 but are still far from being investigated to their full potential.
In this review, we will try to concentrate on these three families of well-defined polymers and highlight their potential applications in the biomedical field. Fig. 1 gives a general overview of the polymers and structures that we have considered for this review.
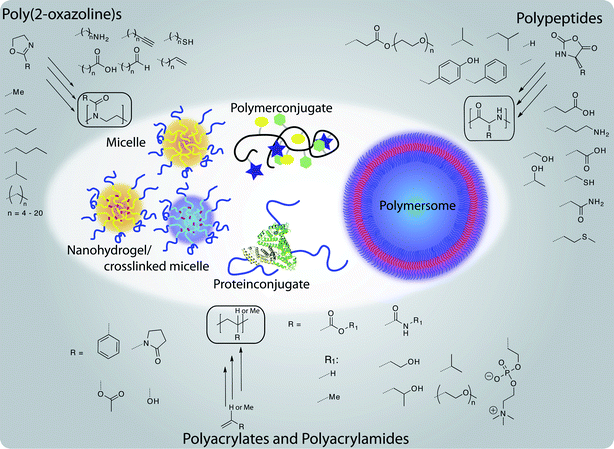 | ||
| Fig. 1 Well-defined polymer architectures accessible from non-PEG polymers for therapeutic applications: a brief overview about structural variety of polymers and resulting structures discussed in this review. | ||
Additionally we also aim to review the data and information that have been obtained in recent years about structure–property relationships of biomedical relevant polymers and their behaviour in in vitro as well as in vivo models. Due to the tremendous advances of synthetic possibilities, a great number of defined polymer architectures are accessible. Although polymer chemists have now various tools to synthesise defined polymers, it remains to be fully elucidated what is the influence of polymeric design on their interaction with biological entities, both on the cellular and on the whole organism level and how we could take profit of this behaviour in selected medical applications.
2. Defined polymeric structures for biomedical application
2.1 Defining definition
The authors of this review and many other researchers are emphasizing more and more the importance of well-defined systems. Characterisation of polymers and their aggregates is a fundamental issue and there are many excellent reviews and books point out problems and potential pitfalls.34–38 Particular care has to be taken when employing commercial analytical systems are commercially available, in which a computer program tries to “guess” the appropriate parameters for a proper “quantitative” evaluation and supplies “values”. It requires a skilled operator to verify that the assumptions applied are indeed reasonable as a computer program typically lacks this ability. For polymers, it appears that there is a broad consensus that the dispersity (Đ, formerly also polydispersity (index) PD(I)), defined as the ratio of weight average molar mass (Mw) and the number average molar mass (Mn) should be below 1.2 and as close to 1.0 as possible. We would like to shortly demonstrate the influence of the dispersity on the effective distributions of polymers in the size. Small (≤5–10 kg mol−1) and water soluble polymers such as PEG, poly(2-methyl-2-oxazoline) (MeOx) or poly(2-hydroxypropylacrylamide) (PHMPA) are cleared rapidly via the kidneys, because the hydrodynamic radius or diameter is below the renal filtration threshold (approx. 3.8 nm). However, as the hydrodynamic volume increases, the polymers will be retained more and more in circulation until eventually the polymers are not filtered anymore in the glomeruli (pore size approx. 7 nm). Typically serum albumin (M = 66 kg mol−1) is used to define the upper limit for renal filtration but it is important to keep in mind that human albumin is a rather compact, negatively charged, globular protein. We are well aware that particle characteristics, such as size, surface charge, surface polarity and mechanical properties influence their in vitro and in vivo fate.39,40 In addition, the secondary or tertiary structure of proteins reduces the structural freedom dramatically, which is not the case for a random coil polymer or a non-crosslinked polymer aggregate. Especially random polymer coils have the chance to undergo some kind of reptation and can be cleared from the organism even though the hydrodynamic radius is larger than the glomeruli pore size. In respect to this, we would like to point out that neither the molecular weight nor the hydrodynamic radius are ideal measures. Furthermore, it should be noted that reducing the renal excretion to a mere size effect is an oversimplification and other excretion pathways have to be regarded. However, we believe it is worthwhile to have a look on the effects of the unavoidable molar mass distribution of materials when using a traditional polymerisation approach, although it creates a simplified image of reality. Material obtained via a so-called living polymerisation should yield a Poisson-type distribution. At sufficiently high degrees of polymerisation (approx. 30) this can also be described by a Gaussian normal distribution, of which the variance and thus, the resulting dispersity can be conveniently adjusted.41 The normal distributions that correspond to polymers with number average molar mass of 40 kg mol−1 and dispersities of 2, 1.5, 1.2 1.1, 1.04 and 1.01 are displayed in Fig. 2. It is obvious from this representation that distributions with Đ > 1.1 do have significant contributions of masses above 60 kg mol−1. As a polymer chemist one is typically content to achieve dispersities around 1.2. Judging from the expected distribution in such a case, the serum half-life and tissue distribution must be expected to be far from homogenous. The calculated relative molar fractions as well as mass fractions of polymers with Mn = 40 kg mol−1 and Đ (1.5, 1.2 and 1.04) are listed in Table 1. It appears that while for very narrow distributions (1.04) the amount of polymer above the renal excretion limits remains very low (approx. 1%) already a narrow Đ of 1.1 yields 10 wt% of polymer above 60 kg mol−1. At “extreme” values of Đ = 1.5 already half (49%) of the mass of the administered polymer would be above the excretion limit. On the contrary, when a Mn of 25 kg mol−1 is assumed, dispersities of up to 1.2 result in less than 1% of non-excretable material and even at Đ = 1.5 only 2 wt% of the polymer are above 60 kg mol−1. We would like to emphasize that these values relate to model calculations with perfectly symmetrical Gaussian normal distributions and a hypothetical excretion limit of 60 kg mol−1. Moreover, a few polymers which are discussed as biomaterials have biodegradable backbones (e.g.polypeptides, polyesters) and into non-degradable polymers biodegradable segments can be introduced.42,43,44 Therefore, ultimately, such materials are degraded into excretable fragments. However, on the timeframe of pharmacokinetics, we think that such considerations are helpful for the design of appropriate macromolecular carriers for parenteral applications as well as for the understanding in vitro and in vivo experiments.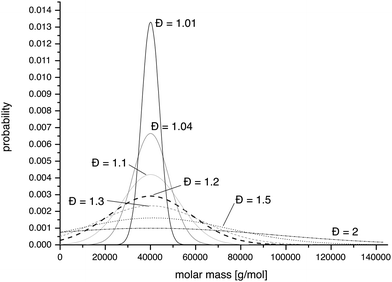 | ||
| Fig. 2 Representation of theoretical Gaussian distributions of PHPMA with a degree of polymerisation of 300 (Mn = 40 kg mol−1) with a variation in the dispersity from 1.01, 1.04, 1.1, 1.2, 1.5 to 2. | ||
| Dispersity | Interval i | ∑(ni)/∑(n) | ∑(niMi)/∑(nM) |
|---|---|---|---|
| Đ = 1.04 | 35–45 kg mol−1 | 47 | 47 |
| 20–60 kg mol−1 | 99 | 99 | |
| >60 kg mol−1 | 1 | 1 | |
| Đ = 1.2 | 35–45 kg mol−1 | 22 | 22 |
| 20–60 kg mol−1 | 72 | 74 | |
| >60 kg mol−1 | 14 | 24 | |
| Đ = 1.5 | 35–45 kg mol−1 | 12 | 12 |
| 20–60 kg mol−1 | 46 | 49 | |
| >60 kg mol−1 | 25 | 49 |
2.2 Defined polypeptides and polypeptide hybrids
On the other hand, synthetic polypeptides were already described by Leuchs in 1906 although their polymeric nature was not acknowledged at that time.48 Many researchers have devoted their research to synthetic polypeptides, in particular since the 1950’s, but poor results have been achieved regarding polymerisation kinetics, end-group analysis or molar mass in particular with more complex systems, such as, block copolypeptides, star-like polypeptides or bottle-brush polymers. This has several reasons. First, it is relatively difficult to obtain the monomers, amino acidN-carboxyanhydrides (NCA), in sufficiently high purity. Second, the monomers are highly reactive and in some cases cannot be stored over prolonged periods of time and their decomposition products themselves can initiate the NCA polymerisation. Third, the classical polymerisation does not necessarily follow a single mechanism. Instead, a multitude of interchangeable pathways exist which, in addition to physicochemical factors give rise to broad, sometimes multimodal molecular weight distribution and in particular poor control over chain termini and length (Fig. 3). Without going into too much detail (which can be found in excellent books and reviews49,50), several aspects are notable. First, using primary amines for initiation of NCA polymerisation is the method of choice as they typically give a rapid initiation as compared to propagation, an important prerequisite for defined polymers. Secondary or tertiary amines, alcoholates or most other nucleophiles will either give slow initiation with respect to propagation or initiation via the activated monomer mechanism (AMM) as opposed to the normal amine mechanism (NAM) expected for initiation by a nucleophile. Unfortunately, the growing NAM initiated polypeptide chain does not necessarily stick to this mechanism but may initiate AMM at any point during polymerisation while any AMM initiated polymer chain can simultaneously propagate via the NAM mechanism. In addition, NCA anions are well known to be able to rearrange into α-isocyanatocarboxylates. To make the situation worse, intermediate carbamates can also lead to a nucleophilic attack to NCAs. On top of all this, most oligopeptides tend to form secondary structures even at very low degrees of polymerisation, most notably α-helices and β-sheets. Both forms differ strongly in solubility and reactivity towards further polymerisation. To conclude, classic NCA polymerisation tends to be very problematic, even when initiated by primary amines.
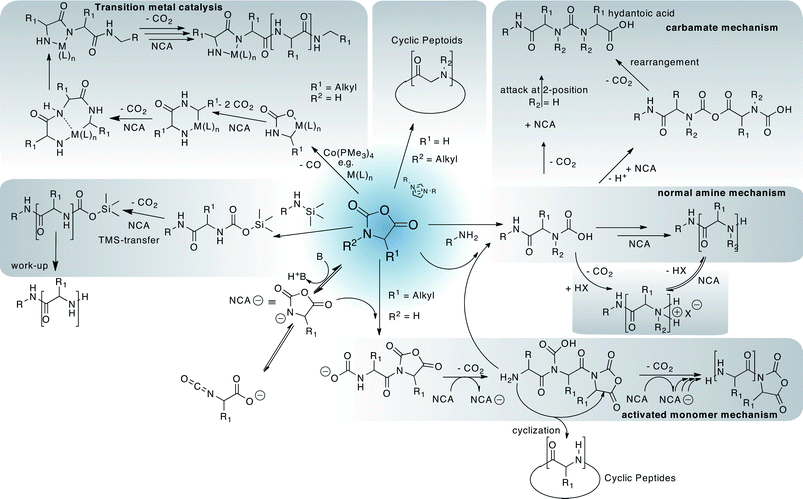 | ||
| Fig. 3 A simplified selection of possible polymerisation approaches and side reactions during polypeptide synthesis from amino acidN-carboxyanhydrides (NCAs). | ||
In the late 1990's, Deming was the first to describe the synthesis of defined polypeptides in a well-controlled manner using transition metal catalysts.32 This approach has been very successful for the preparation of highly defined and complex polypeptide architectures but has two potential shortcomings. First, no specific initiator function can be introduced into the polymer and second, the need for a metal catalyst. Therefore, the run for defined polypeptides is still ongoing and a large number of researchers dedicated their efforts to find alternative ways towards well-defined polypeptidic systems. Hadjichristidis and co-workers reported on the use of highly purified monomers, solvents and reagents and high vacuum techniques.50 While this approach allows the preparation of very large polypeptides with good definition, it remains to be seen whether it will become a common approach, as it is very challenging from the technological standpoint. Interestingly, these results suggest that all the above-mentioned potential side reactions are impurity related. In contrast, Dimitrov and Schlaad introduced a very facile and diametrically opposed method.51 It is proposed that by the use of protonated amine initiators (i.e. addition of stoichiometric amounts of HCl), side reactions and alternative polymerisation routes are strongly reduced. Similar to controlled radical polymerisation techniques, the nucleophilic amine terminus is transferred into a dormant (i.e. protonated) state. Thus, block copolymers and synthetic peptide hybrids are available using a relatively easy method. What is in particular interesting about this method is that researchers were emphasizing for decades that removal of HCl, the most common impurity from Fuchs–Farthing NCA synthesis, is crucial for successful NCA polymerisation, also because chloride has been described as an initiator of NCA polymerisation.49 More recently, Chen and co-workers have reported on the use of silylated amine initiators, which allow the preparation of defined polypeptides.52 Since the trimethylsilyl residue is present at the polymer terminus, control over the polymerisation is retained. Importantly, in this approach the polymerisation is not slowed down as the authors describe quantitative polymerisation yields (degree of polymerisation ≤ 300) at room temperature within 24 h or less under atmospheric pressure.53 In contrast, the protonated amines in Schlaad's approach lead to a much slower propagation. Here elevated temperatures (40–80 °C) were applied and the polymerisation proceeded for several days.51,54 In contrast to all these approaches, Scholz and Vayaboury are interfering with the aforementioned formation of secondary structures and obtain well-defined polypeptides. It was found that the definition of the polypeptides increased markedly no matter whether macroinitiators (PEG-NH2) or low molar mass initiators (hexylamine) are used.55 Vayaboury et al. also reported that by reducing the polymerisation temperature from ambient temperature to 0 °C, a dramatic increase of amine terminated polymer chains could be obtained as was shown by non-aqueous capillary electrophoresis. Unfortunately no values for the dispersity of the materials obtained by this method have been reported.56 Also, reaction times of a week may not be acceptable for common applications.
Tansey and coworkers reported the synthesis of a branched polyglutamic acid based on a poly(ethylene imine) (PEI) core, modified the polypeptide end groups with a targeting ligand (folate) and evaluated the cellular uptake of those systems.70 One issue of the use of PEI as an initiator is the combination of primary, secondary and tertiary amines. While the primary amines are known to initiate the NAM, tertiary ones enable the AMM mechanism. Furthermore the initiation rates of primary, secondary and tertiary amines are different. These facts lead to a reduced control over the polymerisation yielding less defined systems (branched as well as linear polypeptides) as well as a diminished molecular weight control.49
Lu et al. reported recently on an interesting approach to obtain well-defined polypeptide brushes via combination of two controllable polymerisation mechanisms, the ring-opening metathesis polymerisation (ROMP) of norbornene derivatives (backbone) and the TMS initiated NCA polymerisation of L -glutamic acid, L-lysine and L-leucine (side chains).61 In a one-pot synthesis, they were able to obtain very well-defined polypeptide brushes differing in chain length and side chain structure. It was shown that both polymerisations were very well controlled and the final products had dispersities well below 1.2 and polymers with molar masses as high as 500 kg mol−1 could be achieved. Kinetic investigations showed that side chain NCA polymerisation was efficient, at least when only approx. every fourth monomer along the backbone served as an initiator. Whether this is enough to obtain rod-like molecular brushes remains to be elucidated. Although a norbornene backbone would be a problematic choice for any biological application this proof of principle is very important.61 Nevertheless, such excellent control over the backbone and side chain lengths allows the preparation of a great variety of polymer structures from the same monomers. The large pool of natural and non-natural amino acids offers a multitude of possibilities to tune polymer structure and properties. Thus, synthetic polypeptides remain a very promising field of research leading to the investigation of detailed structure–property relationships and development of peptide based polymer therapeutics. Not only the synthesis, but also the characterisation of complex systems remains challenging. Beside end group analysis, determination of branching parameters is required. One possibility is the incorporation of a cleavable position within the initiating site. This approach allows the controlled decomposition of the complex architectures and enables the characterisation of the linear polymer. Since polypeptides are backbone-degradable, too, this cleavage must ensure that the polymer itself remains unchanged.
Cyclic polymers are interesting alternatives to linear ones, albeit more in an academic point of view for the moment.71 Cyclic polypeptides have been described as a side product from base initiated or thermal polymerisation of NCA monomers.72 More recently, however, a new synthetic approach with cyclic polypeptoids as main product has been reported. When using N-heterocyclic carbenes (NHC) as initiators, Guo and Zhang found that cyclic (block) copolypeptoids were the predominant product.73 While this synthetic approach will be limited to N-substituted NCAs, cyclic polymer are certainly intriguing materials to study structure–property relationships in comparison to their linear analogues.
It is well known that the size and steric demand of (polymer) amphiphiles have a significant effect on the nature of aggregates formed in aqueous solution. Simple spherical micelles, polymersomes but also nanorods and nanotubes can be formed. For example, Kimura and co-workers observed that the morphology of the molecular assemblies was tunable by suitable molecular design of the hydrophobic block, selection of the chain length of the hydrophilic block and processing.65
One, potentially significant problem of polypeptides should always be kept in mind. Peptide fragments are a fundamental basis of immune response. Especially when different amino acids are incorporated into a polypeptide, immunogenicity must be anticipated. This significantly limits the molecular tool kit given by the amino acids as many if not most possible combination would lead to immunotoxicity. This problem may also occur when non-immunogenic polypeptides such as PLGA are combined with drugs or other synthetic polymers such as PEG. Such issues should be addressed when designing and developing polypeptide based materials. On the other hand specific modulation and interference with the immune system by designed and defined polypeptides give the chance to develop new polypeptide based drugs and adjuvants.74
For a much more detailed overview on the chemistry and application of polypeptides from NCA polymerisation, the reader is referred to excellent reviews by Kricheldorf,72 Deming74 and Hadjichristidis and colleagues.75
2.3. Poly(2-oxazoline)s, the flexible pseudo-peptides
Previously, poly(2-oxazoline)s (POx) or poly(N-acetylethylenimine) were mainly of interest for researchers in the drug delivery field as a convenient source for linear poly(ethylene imine) used in gene delivery (non-viral transfection vector). However, more recently, several research groups divert considerable efforts towards the use of POx as a versatile building block for drug delivery systems. POx can be regarded as pseudo-polypeptides as each repeating unit contains a peptide bond, albeit in the side chain instead of within the main chain. They are prepared by living cationic ring opening polymerisation (LCROP) from 2-oxazolines and are available with a large array of reactive/functional (protected) and non-reactive side chains (Fig. 1, 4). Several monomers are commercially available (e.g.2-methyl-, 2-ethyl- and 2-phenyl-2-oxazoline), but the majority has to be synthesised. In most cases, this is possible by straightforward procedures from readily available commercial sources, typically nitriles or carboxylic acids.76 The polymerisation can be initiated by, among others,76 alkylhalogens, -tosylates or -triflates and is surprisingly robust as compared to other living polymerisations.77 Again, fast initiation in comparison to propagation is important. In this respect, triflates (and to a lesser extend tosylates) are the preferred initiators.76 The polymerisation is regarded as a living one, although side reactions cannot categorically be ruled out.78 Termination can occur by nucleophilic impurities or be achieved by addition of N- (e.g.piperazine derivatives79,80), O- (water/carboxylates81) or S-nucleophiles (thiols/thioacetate82,83). Considering that tosylates and triflates are readily prepared from alcohols, both termini of POx are easily functionalised with a large variety of functional or reactive moieties. In addition, most monomers can be quantitatively converted into the so-called initiator salt by reaction with stoichiometric amounts of triflate/tosylate. These can be isolated and used for initiation at a later time.80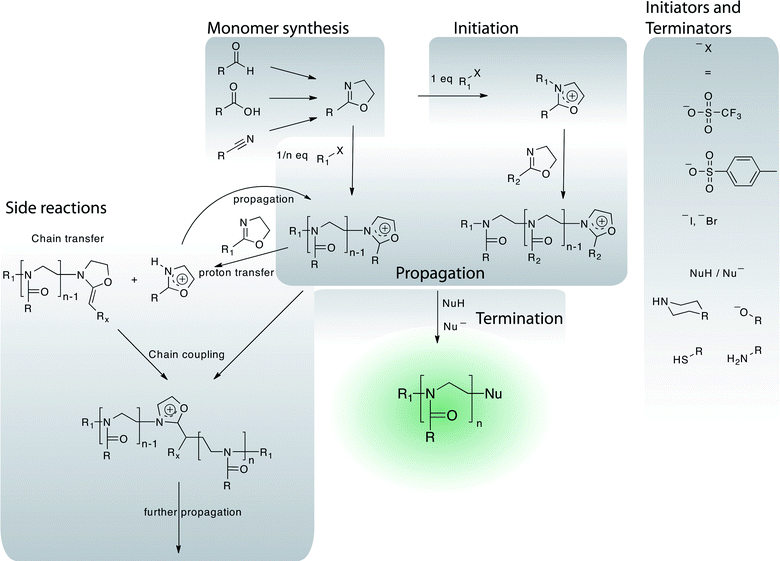 | ||
| Fig. 4 Overview of the chemistry of the polymerisation of 2-oxazolines including monomer synthesis, initiation, propagation and termination reaction. A selection of possible side reactions is outlined. | ||
Depending on the nature of the pending side chains, these polymers are hydrophilic (e.g.methyl (MeOx)), show amphiphilicity84 and thermoresponsiveness (e.g.ethyl (EtOx), n- and iso-propyl) or are hydrophobic (e.g.butyl, nonyl, phenyl)/fluorophilic (e.g. fluorinated phenyl76,85). For reactive side chains, aldehyde,86alkyne,80,87 carboxyl,88thiol,89amine,90,91hydroxyl,88azide87 and others have been described and used for polymer analog modifications. This variety is important as it offers the great potential for the preparation of multi- and polyvalent polymer conjugates for therapeutic applications.
The polymer microstructure is of importance as it will strongly influence aggregation behavior and aggregate stability which in turn will affect the interactions of aggregates with amphiphilic compounds in the blood stream (proteinse.g., serum albumin) or biological barriers (cell membranes).92
Also the polymer architecture is an important parameter for the pharmacokinetic behavior in vivo. Jordan and co-workers recently introduced defined star-like POx as well as molecular brushes by the use of pluri- and polytriflate initiators.93–95 In contrast to halogen-based multi-initiators,96 these give a much faster (and quantitative) initiation rate in comparison to the relatively slow polymerisation. Unfortunately, no pharmocokinetic data are available on these polymers up to date.
In addition, POx have been combined with a great variety of other polymers with potential for biocompatible materials for therapeutic applications, including polyesters97 and polypeptides.98–101
The formation of flexible secondary structures by chiral POx has been recently reported by Hoogenboom and Schubert, represents a promising tool for the extension of the modular kit; the POx system represents and opens the door to new, potentially biocompatible materials with interesting properties. However, the investigated chiral POx are insoluble in most solvents, resembling the behavior of α-helical oligo- and polypeptides which might actually limit their applicability.102–104 At this point, these structures seem to be rather transient with a low persistence length, but the proof of principle is likely to trigger more detailed investigations.
Lipopolymers of POx are easily accessible using lipid initiators. Zalipsky and co-workers used POx-based lipopolymers for the preparation of liposomes and showed that hydrophilic POx can prolong the circulation of coated liposomes similar to PEG.105 In contrast, low molar mass hydrophilic POx are readily excreted via the kidneys and show no unspecific accumulation in any organ.106 Jordan and co-workers used such lipopolymers for the preparation of polymer supported artificial membranes.107,108 It was shown that large transmembrane receptors such as integrins can be integrated and studied in such systems.108 Surprisingly, despite the very promising data elucidating the stealth effect of hydrophilic POx,105 no studies on micelles/aggregates/liposomes comprising POx based lipopolymers for drug delivery have been published up to date. However, as of now, a lack of detailed biological evaluations of POx based systems is apparent, although it has been reported that POx show no adverse effects in rodents after injection of up to 2 g kg−1.109
POx–enzyme conjugates have been known for decades as alternatives for PEG-conjugates and it is known that POx conjugation (sometimes termed POxylation, POXAylation or POzylation) can solubilize enzymes in organic media and helps to retain enzyme activity therein. In an early work, the presence of water along the POx backbone was suggested to cause the enhanced enzymatic activity in benzene as compared to PEG.110 The living cationic termini of POx have also been used to directly attach a bioactive peptide.111
A POx based copolymer system has also been discussed for the preparation of vaccines. However, the authors chose a synthetic route which leads to extremely undefined polymers, therefore these carriers will not be discussed in more detail.91,112
Despite the rich side chain chemistry that would allow for the attachment of bioactive compounds, very few reports can be found in the literature using this potential strategic advantage of POx over PEG. Luxenhofer et al. used POx with pending alkyne moieties for the attachment of RGD peptides along the backbone while an amine terminus was used for the attachment of a radionuclide chelator.113 Similarly, the reaction between pending aldehydes and amino-oxy bearing peptides was used for the preparation of polymer–peptide conjugates.114 More recently, Schlaad and co-workers used the reaction between unsaturated POx side chains and thiols for the attachment of sugars which could be also used as targeting moieties in the future.115 Manzenrieder et al. recently described the decoration of a viral coat protein with PMeOx and PEtOx chains via click chemistry. Such, well-defined and very stable protein nanocontainers may serve as interesting drug delivery vehicles in the future.116
Besides these covalent approaches, several non-covalent formulations have been reported. In a series of papers spanning the 1990s, Maeda and co-workers investigated the formation of nanoparticles with enzymes such as horseradish peroxidase, catalase and lipases in the presence of amphiphilic block copolymers of POx, typically comprising 2-butyl-2-oxazoline in the hydrophobic domain.117–121Enzyme activity was not diminished, on the contrary, lipase activities were even enhanced in aqueous environment, presumably by increasing the local concentration of lipase substrates.118,120 Similarly, enzymatic activity of the enzyme–POx particles was increased in organic solvents. These systems were applied for the preparation of a biosensor.121 In the same manner, the interaction of such POx amphiphilic block copolymers with human serum albumin (HSA) was studied.122 Surprisingly, the studied amphiphilic block copolymers did not interact with HSA through the hydrophobic moieties but rather with the hydrophilic corona, in this study PMeOx. Although the amount of HSA interacting with the POx micelles was found to be rather low, this is particularly interesting since a more recent study suggests that PMeOx exhibits very little interaction with other proteins.123
The groups of Meier and Montemagno have been working over the last decade with copolymers of MeOx or EtOx and poly(dimethylsiloxane).124–130 Although using the route applied by both groups defined polymers are not necessarily obtained, it was shown that bioactive functionalities can be incorporated into the polymersomes formed by such block copolymers. However, whether such polymers can in fact be useful in a biological setting remains to be elucidated.
Another point of interest in water-soluble polymers is the phenomenon of a change in water solubility in dependence of temperature. The lower critical solution temperature (LCST) can be observed for the majority of water-soluble polymers. Above a certain temperature, the polymers become insoluble and precipitate. When used in networks such as hydrogels, the hydrogels collapse. Two points are especially of importance for applications of this phenomenon, (i) being able to tune the temperature of the phase transition and (ii) obtaining materials with rapid and sharp transition when the respective temperature is reached. For specific applications, reversibility and lack of a hysteresis are also of importance. As mentioned before, the side chain of POx strongly influences their properties, also their water solubility. With methyl substituents, no LCST is observed and the polymer is highly water soluble, in fact hygroscopic. Also PEtOx are very soluble in water, however, this polymer already shows a LCST of 60–70 °C, depending on the polymer architecture and degree of polymerisation. POx with isopropyl and n-propyl side chains show LCSTs of ∼40 °C and 25 °C, respectively, while poly(2-butyl-2-oxazoline) (PBuOx) is not anymore water soluble. Further tuning of the LCST can be achieved by two means, copolymerisation of different monomers and modification of polymer termini.131–133 Thus, LCST values covering almost the entire range of liquid water have been achieved. The low dispersity of the polymers is of great importance also in this context. Since the LCST of polymers can depend, among other factors, on the molar mass, samples with a higher dispersity will naturally contain species with differing thermal behavior, thus broadening the transition. In order to achieve a rapid and complete phase transition in a narrow temperature interval, high polymer definition (i.e. low dispersity) is favorable.
Additionally, polymer analog modification of unsaturated side chains with hydrophilic and hydrophobic moieties also allowed LCST modification over a wide range.134 Especially the latter method is interesting in the context of polymer conjugates for therapeutic applications. Bioactives that are covalently attached to water soluble polymers are in the vast majority of cases hydrophobic. Therefore, the physicochemical properties of such conjugates need to be studied at physiological conditions.
For a more detailed and very recent overview on the potentials of POx for other applications, the interested reader is referred to a recent review by Hoogenboom.33
2.4 Defined polymers obtained by controlled radical polymerisation techniques
The development of controlled radical polymerisation (CRP) techniques, sometimes also termed living radical polymerisation (LRP) techniques, had a tremendous impact on synthetic polymer chemistry. The CRP techniques were developed to reduce termination as well as uncontrolled transfer of radicals, and are divided into three subgroups, which are stable free radical polymerisation (e.g. NMP31), degenerative transfer polymerisation (e.g. RAFT, MADIX) and transition metal-mediated controlled radical polymerisation (e.g.ATRP). Among these, ATRP and RAFT are arguably the most commonly used and most versatile processes. There have been various reviews describing mechanism as well as recent developments of either RAFT30,135 or ATRP.29,136,137 The CRP techniques can be used in the synthesis of complex polymer architectures e.g., (multi) block copolymer, branched polymers or hybrid systems.138–141 During the last few years, some reviews have already focused on the recent advances towards biological application of both techniques.142–145 These detailed and interesting reviews have focused more on the synthesis of new polymers and polymer architectures, but less on biological or medical application of defined systems. In this respect, we would like to point out materials, which can be expected to enrich the pool of building blocks for polymers in biomedical applications.Briefly, ATRP is a means of forming carbon–carbon bond through transition metal catalyst. As the name implies, the atom transfer step is the key step in the reaction and therefore it is responsible for uniform polymer chain growth. The uniform polymer chain growth leading to polymers with rather low dispersities is mainly related to the transition metal based catalyst. This catalyst provides an equilibrium between active polymer propagating the polymerisation and its inactive form, which is commonly described as the dormant species. Since the dormant state of the polymer is under appropriate conditions greatly preferred in this equilibrium, the concentration of propagating radicals is constantly low. Thus, side reactions, e.g. termination and recombination, are effectively suppressed and control over molecular weights can be achieved.
The ATRP allows the polymerisation of many functional groups including allyl, amino, epoxy and hydroxy groups present in either the monomer or the initiator. ATRP methods are also advantageous due to an easy preparation, commercially available and inexpensive catalysts (copper complexes), pyridine based ligands and initiators (alkyl halides). Only the copper content may influence biological systems even though it is usually kept below the upper limit of copper approved for medical application.
In contrast to ATRP, the RAFT polymerisation technique does not require any metal catalyst. Instead, thiocarbonylthio compounds, such as dithioesters, dithiocarbamates, trithiocarbonates, and xanthates (MADIX) are employed in order to mediate the polymerisationvia a reversible chain-transfer process. These reagents are called chain transfer agents (CTA). The mechanism itself is complex. It is based on two chain-transfer and two chain-propagation equilibria establish control over the radical polymerisation. In this process, a growing polymer chain reacts with the CTA yielding an intermediate radical. Due to the chemical structure of the CTA it can fragment in two ways. This leads to a new chain transfer agent and a free radical, which can propagate the polymerisation. Thus, the propagation probability is equally distributed over all polymer chains, which is the reason for narrowly distributed polymers. Furthermore, the ongoing transfer of radicals between growing and thiocarbonyl thio terminated chain enables a polymerisation with reduced concentration of radicals. In respect to this, side reactions are effectively reduced. In addition, it is important to point out that the average chain length is proportional to the concentration of the CTA as well as to the monomer conversion.
Some disadvantages of the RAFT polymerisation have to be kept in mind. First careful choice of chain transfer agent, reaction conditions and monomer is required to achieve control over the polymerisation. Second, the (macro) thiocarbonyl thio group of the (macro) CTA can undergo various side reactions, which may create the issue of end group attributed in vitro toxicity.146,147 On the other hand, the reactivity of the thiocarbonyl thio group can be used to modify the end groups of the synthesised polymer afterwards.148 For example the CTA can be oxidised or reduced149 as well as decomposed thermally,150 by an excess of radicals151 or by nucleophiles152e.g., amines153 or hydroxide ions.154 Those reactions can be used to attach bioactive agents.155,156 Regarding those end groups it is important to keep in mind that every CRP method has still characteristics of a radical polymerisation. Thus, the end group integrity cannot be complete. Side reactions can be reduced to a certain extent, but never fully eliminated. A brief discussion of possible side reactions can be found in the early review of Moad et al.135 This fact implements that every modification of end groups or grafting from approaches need careful purification to eliminate by-products. Especially in the field of protein modification the separation of covalently bound and weakly adsorbed polymer has to be ensured. In addition free thiol units within a protein may interfere with the CRP and act as a chain transfer agent leading to less defined conjugates.
During the last few years not only polymerisation methods have improved tremendously. In addition, a variety of novel monomers yielding biocompatible polymers were investigated. The number of these systems is rather high and a detailed description of developments is beyond the scope of this review. Here, we would like to focus on some examples of polymeric materials, which already have been applied to biological investigations. Additionally, we would like to summarize useful synthetic approaches, which allow highly functional and biocompatible polymeric structures, e.g. the post-polymerisation modification of reactive polymer precursors.157–159
Many new polymers belong to the group of poly(meth)acrylates or poly(meth)acrylamides. Among these monomers the group of (meth)acrylates bearing oligoethylene glycol side chains (OEGMA), e.g.diethylene glycol methacrylate (DEGMA) or polyethylene glycol methacrylate (PEGMA) have seen an increasing interest. These systems have rather interesting properties, such as a high solubility in water, a non-immunogenic and non-toxic character, a lower critical solution temperature (LCST) and enhanced blood circulation times.160–164 The LCST can be nicely tuned by copolymerisation of both monomers. It was reported by Lutz and Hoth that the LCST can be adjusted from 26 °C to 90 °C by changing the ratio of OEGMA to DEGMA units in the copolymer.165
These oligoethylene glycol based monomers have been applied to ATRP as well as RAFT polymerisation leading to well defined homo, random, block or star (co)polymers.161 Additionally, block copolymers prepared from these monomers have shown interesting superstructure formation in solution. The biomedical application of micelles166,167 and polymersomes168 has been reported during the last few years. Ethylene oxide based systems appear to offer various advantages, as PEG has already achieved clinical approval and entered the market24 and their safety is easily postulated writing proposals and manuscripts. But it has to be kept in mind even though the material might appear comparable, the physicochemical and biological properties of these (meth)acrylates are different. The PEG side chains are usually rather short (2–9 units) in order to achieve material suitable for biomedical applications. Ryan et al. reported that linear PEG grafted onto salmon calcitonin enhances the serum half-life, while comb-shaped PEG displayed increasing resistance of the protein against intestinal enzymes, liver homogenate and serum.170 Additionally Gao et al. reported also improved pharmacokinetics by N-terminal conjugation of POEGMA to myoglobin.171 Cytotoxicity was investigated in various cell lines, e.g. Caco-2, HT29-MTX-E12 or HepG2, ensuring nontoxic behaviour up to 5 mg mL−1.170,172
As a main advantage of these polymers over PEG the possibility of copolymerisation with other reactive monomers should be mentioned. Thus, multifunctional systems can be synthesised overcoming the problem of the α,ω functionality of PEG. On the other hand, monomers with a relatively large molar mass inevitably give rise to broader distributions (if Poisson distribution applies), in particular at low degrees of polymerisation (<20). As for any new materials, these systems have to be investigated in detail, before they can be considered as a substitute for PEG.
Many other interesting polymer systems are based on poly(2-(meth)acryloyloxyethyl phosphorylcholine)s (PMPC)s. The monomer structure is highly bio-inspired, because the side chain contains the head group of the natural phospholipid phosphatidylethanolamine ensuring high biocompatibility. It was polymerised by ATRP173–175 as well as RAFT176–179 yielding various well defined polymer architectures, micelles174,180 and polymersomes.181–183 Polymersomes have been applied to study diffusion across oral epithelium and were used as transfection agents by Battaglia et al.183 with pronounced cellular uptake as well as non-toxic behaviour. In addition, PMPC was used for protein conjugation by Lewis and coworkers.184 A reduced tissue migration compared to PEG–protein conjugates of the same hydrodynamic volume was observed. Thus, an improved depot effect in the tissue as well as subsequent longer elimination half-life may lead to improved pharmacokinetics. These findings underline the potential of PMPC based polymeric systems for further medical application.
Another group of polymers having a potential for medical application are glycopolymers, which have been investigated by various groups regarding synthesis, physicochemical properties as well as first biological evaluations.185–189 In nature glycosides or glycopeptides are the key to various processes in cell–cell interactions. The glycocalyx, the outer, highly glycosylated, cellular envelope, is involved among others, in inflammations, viral infections, fertilisation and signal transmission. In this respect, glycopolymers can be expected to provide interesting properties for medical applications,190e.g. immunotherapy of cancer or treatment of auto-inflammatory diseases.191 This natural glyco-code is highly complex and therefore structures mimicking or interactions with it are highly complex. For example the total synthesis of siaLex includes at least 26 steps192 yielding a pure P-selectin glycoprotein ligand 1 (PSGL-1), which plays a major role in the inflammatory cascade193 and may be a useful tool in diagnostics and treatment of autoinflammatory diseases. For such highly complex structures, mimicking agents are desirable. In this respect, well-defined glycopolymers or glycoside functionalised polymers would be beneficial.
Deng et al. reported a non-toxic behaviour up to 5 mg mL−1 of a gluconolactone derivate bearing block copolymers in HeLa cells.194 In addition lectin binding experiments were carried out by Granville et al. leading to the interesting result that the protein–carbohydrate binding is completely disrupted when the 6-carbon position is modified.195
Ayres et al. prepared polymer brushes containing sulfonated sugar monomers by ATRP. They compared these systems with unsulfonated analogues in vitro. The sulfonated brushes showed improved blood compatibility in terms of plasma recalcification, clotting times and complement activation.196
Nevertheless immunogenic properties have to be carefully investigated whenever an in vivo application is desired. Glycopolymers may bind to their targets, but the polymer has to provide specificity in vivo when more than one interaction side is available. Why would nature use an exceedingly complex structure, when a simple undefined motive would do the same job?
Beside the above mentioned systems well-established polymers for therapeutical application have been prepared using the new synthetic methods of CRP. During the last few years several groups have applied RAFT or ATRP to the synthesis of functional 2-(hydroxypropyl) methacrylamide (HPMA) based polymers.197–201 Some of these systems will be discussed in detail in the last chapter of this review. In addition the authors would like to refer to recent reviews on HPMA based polymer therapeutics providing a much more detailed description of biomedical applications as well as its future prospective.202–209 These articles point out the advantages as well as disadvantages of HPMA as a monomer in biological or medical application. Furthermore the special issue provides interesting insights into research carried out during the last 30–40 years.
Additionally, we would like to mention post-polymerisation modification methods,157–159 which can be considered as an old but still promising approach to highly functional and complex structures based on well-established polymers. In this approach the final structure is not polymerised directly. Instead a reactive precursor polymer is synthesised, which can be precisely characterised and afterwards easily transferred into a final multifunctional system. Currently the most prominent example of the post-polymerisation modification is the Huisgen [2 + 3] cycloaddition, in which an azide reacts with an alkyne, typically under Cu(I) catalysis, forming a triazole derivative.159 During the last few years the number of publications has enormously grown and detailed description is beyond the scope of this review. As an example, Geng et al. have applied this method to the synthesis of glycopolymers.210,211 These glycopolymers were conjugated to BSA yielding artificial glycopeptides. Thus, the normally inert BSA showed the expected stimulation of the immune system.211
Among reactive polymers for polymer analogue modifications, activated esters offer some advantages. First of all, most of them have proven their potential in synthetic peptide chemistry. Second, they have been already applied to the synthesis of the first polymer–drug conjugates entering clinical trails (PK1 and PK2).212,213 Additionally, various activated esters are known in the literature offering tuneable reactivities.158 Another very intriguing advantage is the possibility to synthesize various acrylate or acrylamide based architectures from one polymer precursor. Thus, copolymerisation parameters can be disregarded and amphiphilic block copolymers can be prepared from well-characterised non-amphiphilic copolymers.
Taking advantage of the activated ester approach, the synthesis of a variety of HPMA based polymers as well as glycopolymers was reported.205–208,214 Additionally, in vitro as well as in vivo studies were carried out.92,215–218 Gibson et al. could demonstrate that HPMA based homopolymers derived from poly(pentafluorophenyl methacrylate) (PPFMA) show comparable cytotoxicity as compared to regularly polymerised HPMA.216 Barz et al. reported on poly(HPMA)-block-poly(lauryl methacrylate) block copolymers with no cytotoxicity at concentration up to 3 mg mL−1.215 Moreover, Herth et al. used similar polymers in preliminary in vivo experiments.217 In this work a new radioactive labeling chemistry for positron emission tomography was introduced, monitoring non-invasively the body distribution of various polymeric architectures. However, most importantly, the reactive precursor strategy offers the chance to synthesize functional systems from one precursor polymer as demonstrated by Barz et al.92 and Brocchini and coworkers.218 An important feature of this approach is that even though the degree of functionalisation changes, the degree of polymerisation remains the same.92 This makes the comparison of polymers with different degrees of functionalisation much more meaningful. These structure–property relationships are essential for a more sophisticated design of polymer therapeutics and are therefore discussed in more detail in the next paragraph.
3. Structure–property relationship: influencing cellular fate of polymer carriers and their cargo
In order to investigate structure–property relationships, the first step is to be able to control the structure as exactly as possible. In case of FRP this is typically not possible. If the copolymerisation parameters are not matched, the composition of the copolymers will change during the polymerisation. Accordingly, polymers obtained by this method suffer in particular from three different distributions: (1) degree of polymerisation distribution, (2) quantitative comonomer distribution and (3) spatial comonomer distribution. Subsequent fractionation is only able to narrow the resulting hydrodynamic radius distribution (which appears to be mainly the molar mass distribution) directly and may or may not influence the other distributions. The latter two problems, however, are very difficult to address by post-polymerisation purification techniques. However, the microstructure, i.e. the distribution of comonomers along the polymer chain has a major influence on the endocytosis in mammalian cells as was recently shown by Barz, Luxenhofer et al. (see Fig. 5).92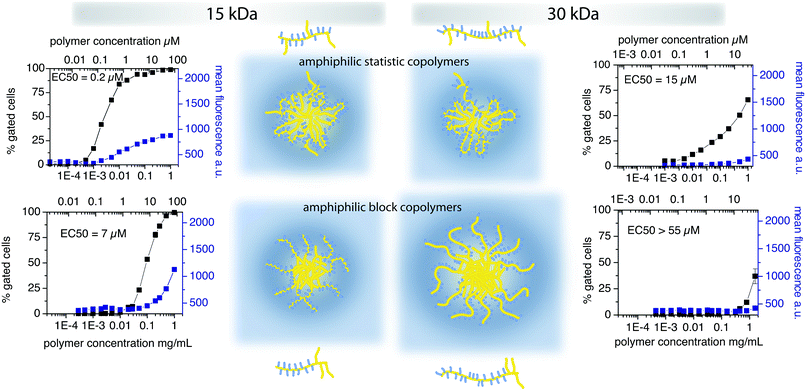 | ||
| Fig. 5 Influence of polymer architecture on cellular uptake kinetics in MDF-7/ADR (human prostate cancer) cells.92 | ||
In contrast to the situation in FRP, polymers obtained via controlled or living polymerisation techniques grow over the whole course of the polymerisation. Thus, the relative comonomer content does differ relatively little in the final polymers obtained by controlled polymerization methods (ideally following a Poisson distribution). In this respect, the CRP are of essential importance in the synthesis of defined polymers on which structure–properties relationships can be discussed. As for the spatial arrangement of the monomers within the resulting polymer chains, differences between CRP and FRP also may not be negligible and, again, may not be addressed by post-polymerisation fractionation. Reactivity differences in FRP (in most cases) are expected to give random copolymers with changing relative monomer content over the course of the polymerisation. Thus, in extreme cases mixtures of different pseudo-homopolymers are obtained. In contrast CRP techniques should yield gradient copolymers in the case of different monomer reactivities (see Fig. 6).
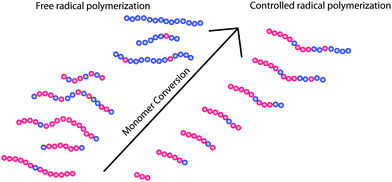 | ||
| Fig. 6 Illustration of the different polymer architectures obtained during free radical polymerisation and controlled radical polymerisation when monomers with different reactivities are used. While FRP will yield pseudo-homopolymers of the more reactive monomer in early stages of the polymerisation, pseudo-homopolymers of the monomer with less reactivity will result at later stages. Thus, the final product will comprise of the different pseudo-homopolymers and random copolymers. In contrast, CRP methods should yield relatively uniformly gradient copolymers. | ||
Additionally, it has to be mentioned that besides the microstructure other physical properties e.g. surface charge and charge density may be interrelated and influence cellular uptake as well as intracellular distribution. However, the influence of those characteristics is more established, especially for engineered nanoparticles. In these cases direct electrostatic interactions appear to be a major determinant for cellular uptake and intracellular distribution.219
However, the intracellular fate of any material taken up by endocytosis will depend strongly on the mechanism of entry.220 However, we have to point out that research in the field of membrane trafficking and intracellular translocation is dynamic220–222 and more detailed knowledge may lead to different interpretation of results. Thus, our picture of cellular uptake of polymeric particles may evolve tremendously during the next decades. Nevertheless, differences in polymer structure will always influence the aggregation in solution and therefore determine the interaction with biological matter.
Sahay et al. reported recently on differential uptake mechanisms of polymer unimers and their micelles, respectively.223 In this study, an amphiphilic triblock copolymer poly(propylene glycol) (PPG) and PEG, Pluronic P85, was investigated. The authors found that while the unimers entered the cellsviacaveolae-mediated endocytosis, the polymer micelles were taken up via a clathrin mediated route. At the same time, it was observed that P85 was able to inhibit caveolae-mediated endocytosis. It should be emphasised that no ligand for specific cellular uptake was employed in this study. The authors hypothesize that the specific interaction with caveolae may be due to perturbation of these highly specialised structures by changing the membrane microviscosity or membrane curvature. The same group recently reported on the endocytosis of nanogels formed by PEG-poly(methacrylate) block copolymers.224 These crosslinked polymer micelles also enter the cellsviacaveolae in a highly specific manner and were then routed to lysosomes. Caveolae mediated endocytosis is highly regulated in epithelial cells and is typically strongly suppressed in cells forming tight junctions. Accordingly, high uptake of drug-loaded nanogels was observed in cancer cells (MCF7/ADR) and sub-confluent MDCK cells. In contrast, when the MDCK cells became confluent and thus, form tight junctions, uptake of the nanogels was practically abolished. Interestingly, Pluronic P85 and the nanogels share a similar PEG-based corona. A more detailed investigation of the cellular uptake and subsequent subcellular distribution of Pluronic P85 in a variety of cells, including neurons and BBMEC were also reported. Interestingly, in this study it was shown that the polymers could bypass the endosome/lysosome pathway reaching the endoplasmic reticulum and the mitochondria.225 This is of significant importance for a number of reasons. First, bypassing the late endosome/lysosome, may avoid or limit the degradation of sensitive payload. Second, the endoplasmic reticulum and the mitochondria are important organelles involved in a large number of diseases. Therefore, direct delivery of drugs to these organelles may be beneficial. Future studies using PEG and non-PEG based materials will hopefully show whether such specific cellular interactions of non-modified hydrophilic polymers are a more general feature that could be used for the facile preparation of materials with specific biological interactions.
In general, it has to be emphasised that these findings clearly point out the key role of aggregate properties, this knowledge is essential for a more detailed understanding of the processes taking place whenever polymeric carriers interact with biological systems.
Kimura and co-workers used amphiphilic polypeptides and polydepsipeptides to obtain self-assembled aggregates in the form of polymer micelles and vesicles, named peptosomes and lactosomes. In both cases PSar served as the hydrophilic polymer. Long circulation times of 48 h and more were reported64 and the RES was successfully avoided. Thus, it was possible to detect tumours in the liver.63 Interestingly, a comparison of aggregates comprising either polypeptide/polypeptoid block copolymer or polypeptoid/poly(L-lactide) block copolymer revealed that the former showed much lower tumour to liver ratios. Both aggregates were of similar size (32 nm vs. 37 nm) but the PSar block length differed somewhat (degree of polymerisation 60 vs. 90). It remains uncertain whether the difference in the in vivo behaviour could be attributed to the aggregate core material or to the minor differences in the hydrophilic corona. Unfortunately no details on the characterisation of the polymers were described so that the influence in the definition of the polymers forming the aggregates cannot be ruled out.
Nemoto et al. demonstrated recently the effect of the dispersity of star-like poly(N,N-dimethylaminopropylacrylamide) (PDMAPAAm) used as non-viral gene delivery systems on the transfection efficiency.226 This work compares the crude polymer with a slightly higher dispersity (Đ = 1.4) and fractions thereof with lower dispersities (Đ = 1.1–1.2). The authors demonstrate that not only the average molar mass, but also the dispersity does have an influence on the transfection efficiency.
In addition, Callahan et al. have investigated the influence of molecular weight and charge of HPMA-based copolymers on their intracellular distribution after cytosolic microinjection.227 Obviously, cytosolic microinjection is not a valid tool to study the subcellular localisation after endocytosis as in the majority of cases materials will enter through some sort of compartmentalised structure. However, it is an interesting tool to study the fate of materials if they enter the cytosol, often very difficult to achieve by itself. The copolymers were synthesised by FRP and dispersities below 1.7 before and 1.2 after fractionation were reported. However, as mentioned above, fractionation cannot solve all dispersity related issues. Nevertheless, the findings are interesting. All copolymers rapidly and evenly diffused throughout the cytoplasmic compartment after microinjection. The smallest copolymer fractions (Mn = 11–15 kDa) also rapidly diffused into the nucleus. The exception to passive intracellular diffusion was the strongly cationic copolymer containing 20% of a quaternary amine in the side chain. This copolymer was found to localize specifically from the cytoplasm to the microtubules. It was proposed that nuclear entry from the cytoplasm was dictated by size-limited passive diffusion through the nuclear pore complexes (NPC), however, small but significant differences in rates of nuclear import were observed for polymers with sizes near the molecular weight exclusion limit of the NPC as a function of the charge and hydrophobicity of the copolymers. Weak bases were found to have the highest nuclear uptake. These findings indicate a pronounced structure–property relationship, but detailed investigations of the aggregates would be highly interesting. Maybe the differences in aggregation would help to gain a deeper insight.
Moreover, Richardson et al. found pronounced differences in the intracellular distribution of dextrin, HPMA and PEG based polymers228 underlining the tremendous influence of the polymer nature on the cellular fate of the aggregate.
In conclusion, structure–property relationships are not only interesting from the academic point of view but they are also of great importance for the development of polymer therapeutics with clinical applications. The rational design of release systems is only possible, if the cellular fate of the carrier is known.
4. Defined polymers in therapy
Besides PEG only a small number of defined polymers have entered clinical research. As discussed before, especially for in vivo applications defined structures are desirable, because the biological interactions will depend directly on the polymer properties. In the following paragraphs we would like to point out defined polymers for specific applications, most of which are still in preclinical studies. Applications for defined polymers are not only limited to oncology.229 The use of polymer therapeutics is a promising approach to tackle various human diseases.230 In respect to this, different approaches from encapsulation to conjugation of drugs into polymer micelles, polymer–drug conjugates or polymer–protein conjugates have been used in therapy (see Fig. 7 and 8).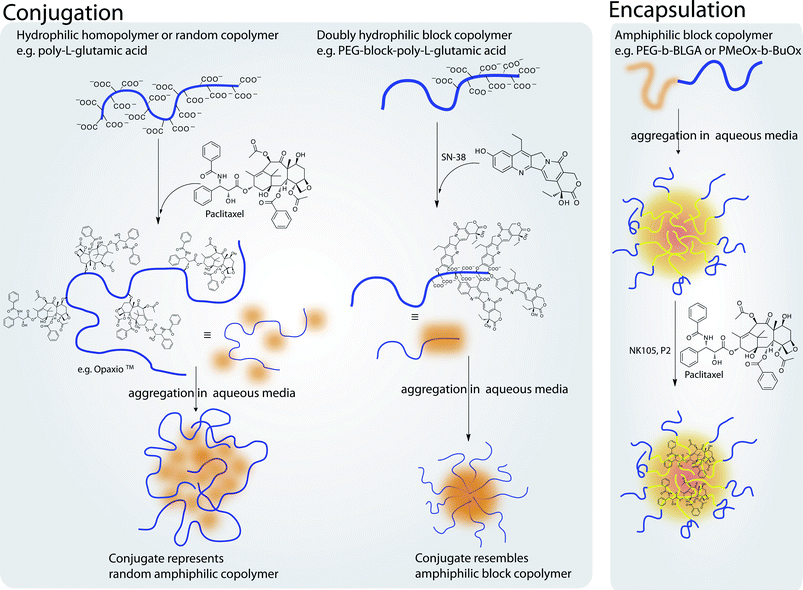 | ||
| Fig. 7 Illustration outlining different approaches of encapsulation and conjugation of drugs into polymer–micelles or polymer–drug conjugates, respectively. Conjugation can be performed to yield structures resembling either random copolymers or block copolymers. In contrast, block copolymers can be used to physically entrap hydrophobic drugs in the micellar core increasing drug solubility. | ||
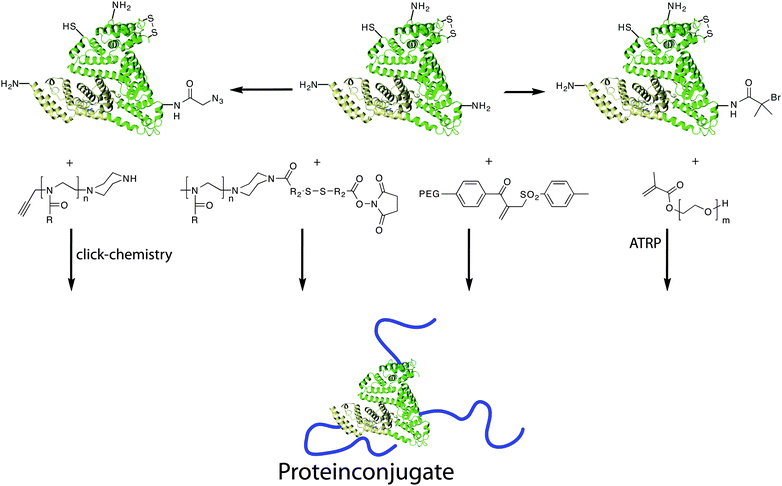 | ||
| Fig. 8 Synthesis of polymer–protein conjugates via different routes. Proteins can be functionalised either with reactive groups that can serve as initiator for e.g.ATRP. | ||
Up to now, polyglutamic acid (PGA) is probably the only biodegradable and water-soluble polymer that can be synthesised or purchased in a relatively well-defined manner (Đ around 1.2–1.4). Thus, it is not surprising that PGA based drug conjugates reached clinical trials. Different classes of drugs have been attached to well-defined PGAs, e.g.anthracyclines, antimetabolites, DNA-binding drugs, paclitaxel or camptothecin.231,232 Among these polymer–drug conjugates Opaxio® (formerly Xyotax, PPX, CT-2103) has been in clinical phase 3 trials for the treatment of non-small cell lung and ovarian carcinoma.233–236 Opaxio® is a polymer–drug conjugate that links paclitaxel through an ester bond to PGA. The release kinetics of the drug is directly related to the degradation of the polymer itself, which is known to be dependent on the enzyme cathepsin B. Therefore, it is important to assess cathepsin B levels in patients. Such, Opaxio® is a representative of personalised medicine in which the patient's particular situation is included in the selection of treatment options.
Kimura and co-workers are using amphiphilic polypeptides and polydepsipeptides (polypeptide-block-polyesters) to obtain self-assembled aggregates in the form of polymer micelles and vesicles, which they term peptosomes and lactosomes. In both cases PSar serves as the hydrophilic polymer. Long circulation times of 48 h and more were reported64 and the RES was successfully avoided similar to PEGylated liposomes of Doxil®. Moreover, labelled PEG could be incorporated into the aqueous core of the polymersomes and colocalised in vivo with the polymersomes 2 days after i.v. injection. It should be noted that in this study the hydrophobic dye was attached to the hydrophilic part of the polymers, which might have an effect on the biodistribution of the respectively decorated micelles and polymersomes. In a similar work, the same group investigated the biodistribution of assemblies of block copolymers of Sar as the first block and L-lactic acid (PLLA) or leucine–aminoisobutyric (Leu–Aib) acid oligomers as the second block. The assemblies were labelled with near IR dyes and the biodistribution assessed in tumour bearing mice. It was found that tumour to liver ratios were considerably above unity peaking above 2. Thus, it was possible to detect tumours in the liver.63 Therefore, all three block copolymers are interesting candidates for further investigations for the delivery of therapeutic molecules. Interestingly, a comparison of aggregates comprising either polypeptide/polypeptoid block copolymer or polypeptoid/poly(L-lactide) block copolymer revealed that the former showed much lower tumour to liver ratios. Both aggregates were of similar size (32 nm vs. 37 nm) but the PSar block length differed somewhat (degree of polymerisation 60 vs. 90). It remains uncertain whether the difference in the in vivo behaviour could be attributed to the aggregate core material or to the minor differences in the hydrophilic corona. Unfortunately no details on the characterisation of the polymers are described so that the influence in the definition of polymers forming the aggregates cannot be ruled out.
Most defined polymer candidates are still in preclinical studies, however one of them, POx can be expected to proceed to clinical studies in a near future.109 In the last decade, POx have seen increasing attention for drug delivery or protein conjugation during the last few years. Jeong and co-workers studied the solubilisation of the highly water-insoluble paclitaxel using well defined (Đ ≤ 1.2) PEtOx-b-poly(ε-caprolactone) block copolymers. They could incorporate up to 7.6 wt% of paclitaxel. The reported micelles only induced very limited hemolysis but some cytotoxicity was observed even at relatively low polymer concentrations (<1 mg mL−1).97
Similar block copolymers comprising PEtOx and poly(L-lactide) were prepared by Hsiue and co-workers. In this account, very low cytotoxicity of the carrier at 10 mg mL−1 was observed. In this study, doxorubicin was used as a bioactive component and 31 wt% of drug loading were reported and the drug was released in a pH dependent manner.99
Hsiue et al. also used POx based polymers for gene delivery. In this account, first pyridyl disulfide terminated PEtOx were prepared. Subsequently, the POx was partially hydrolysed to give a random copolymer of POx and poly(ethylene imine). These cationic-hydrophilic copolymers were subsequently coupled to PEtOx homopolymer to obtain cationic block copolymers structures. Toxicity was relatively low while transfection was similar as compared to linear and branched polyethylene imine.82
The same group recently used a PEtOx–poly(aspartic acid) (PEtOx-b-PAsp) block copolymer for the formulation of amphotericin B (AmB). The carriers showed no cytotoxicity at concentrations of 1 mg mL−1. These polymers were able to incorporate significant amounts of AmB. More importantly, the incorporated AmB was less toxic to mammalian cells as compared to AmB in Fungizone® while its toxicity against Candida albicans was fully preserved. The authors speculated that this might be due to a sustained release of AmB in its monomeric form.101
Lai and co-workers reported on the use of PEtOx-b-poly(D,L-lactide) block copolymers for the delivery of the photosensitizer meta-tetra(hydroxyphenyl)chlorin in tumour bearing mice for photodynamic therapy.237 The particles the authors obtained were loaded with approx. 10% (w/w) of drug and were reported to be 77 nm in size with a very large dispersity (Đ = 0.28). Unfortunately the authors did not mention if the large dispersity is due to a multimodal size distribution or results from a broad but monomodal distribution. The authors showed that while the tumour growth inhibition (HT-29 xenograft) was unaffected by incorporation into micelles, the skin photosensitisation, a major limiting factor of photodynamic therapy, was greatly reduced, in particular when the mice were irradiated only 48 h postinjection.237
Luxenhofer et al. recently reported on the use of POx for formulation of hydrophobic drugs. In this account, di- and triblock copolymers were evaluated which comprise BuOx as the hydrophobic domain and MeOx or EtOx as the hydrophilic part. The authors reported a very high loading capacity for Cyclosporin A, AmB and Paclitaxel. In particular the high solubilisation of Paclitaxel with final formulations with loadings up to 45%wt is striking. The resulting micelles were very small with hydrodynamic radii around 10–20 nm with a narrow size distribution (Đ = 0.04–0.12). The incorporated drug was shown to be active both in vitro and in vivo. Moreover, it was reported that the polymers alone were not cytotoxic at concentrations up to 20 mg mL−1 and showed relatively little complement activationin vitro.238,239
In addition to these non-covalent approaches, several POx–drug conjugates have been described. Veronese and co-workers reported on covalent attachment of trypsin and cytosine arabinose.240 It was shown that the autolysis rate of polymer-conjugated trypsin was comparable between PEGylated and POxylated trypsin. In contrast, the POxylated cytosine arabinose activity was shown to be somewhat lower as compared to its PEGylated counterpart. This, however, was attributed by a somewhat slower drug release from the carrier polymer.
The first commercial enterprise looking into clinical applications of POx is Serina Therapeutics which is currently evaluating POx–drug conjugates for chemotherapy. The POx conjugates are obtained utilizing click chemistry.80,109,113
Another series of conjugates of POx with a therapeutically interesting protein has been recently investigated by Kabanov et al. In this study, horseradish peroxidase (HRP) was conjugated to amphiphilic block copolymers in order to tune the cellular uptake of HRP. It was shown that block copolymers of MeOx and BuOx or EtOx and BuOx are able to increase the cellular uptake of the enzyme in MDCK and Caco-2 cells. In contrast, a hydrophilic MeOx and a random copolymers of EtOx and BuOx did not show this effect.241
Within the group of acrylamide based polymers modern polymerisation chemistry has been applied to the synthesis of polymer therapeutics. Satchi-Fainaro and coworkers have successfully applied the RAFT polymerisation to the synthesis of a copolymer for the treatment of bone neoplasms such as bone metastases and osteosarcoma.242 The copolymer consists of HPMA, TNP-470 and the aminobisphosphonate, alendronate (ALN). TNP-470 is a low molecular weight synthetic analogue of fumagillin able to selectively inhibit angiogenesis and suppress tumour growth. The use of the CRP techniques allowed the synthesis of better-defined polymers (Đ of 1.2 instead of 1.6). The common fractionation of HPMA copolymers could be avoided. Other advantages such as different polymer architectures, as defined end groups as well as grafting from approaches of the RAFT polymerisation have not been used so far.
It is important to note that all CRP techniques offer—in the ideal case—defined end groups as well as access to more complex polymer architectures, e.g.block copolymer. Qiao et al,243 Kirkland-York244 as well as Üzgün245 have successfully used CRP methods to synthesize block copolymers carrying oligonucleotides. These complexes are promising candidates for in vivogene therapy. In both approaches, the nucleotide complexing polycation is shielded by a hydrophilic block, which prevents unspecific interactions and immune responses. Among the nucleotide based systems especially small interfering ribonucleic acid (siRNA)246 is expected to have great therapeutic potential.247–250 Especially, since first indications for RNA interference (RNAi) in cancer patients have been reported by Davis and coworkers using cyclodextrine aggregates as carrier systems.251 Furthermore, Hemmelmann and coworkers have used well-defined poly(HPMA)-co-poly(lauryl methacrylate) polymers to encapsulate and deliver the antidopaminergic drug domperidone® across the blood brain barrier (BBB).252 Although the influence of the polymer remains poorly understood it was clearly demonstrated by applying the rotarod test that this model drug influences the coordinated motor skills of FVB/N-wild type mice. While the drug itself is unable to cross the BBB, but when encapsulated into a copolymer aggregate significant drug related changes in animal behaviour could be demonstrated.
5. Conclusion
The tremendous advances in polymer chemistry and macromolecular engineering brought the accessibility of many new new materials. However, they also allow the preparation of well-known materials in a more defined way. Although the detailed investigation of structure–property relationship using highly defined polymers other than PEG is a relatively young field, it is becoming evident that it is of major importance. This is not at all surprising, as similar trends were already observed for other systems such as highly defined dendrimers. In order to fully use the potential of highly defined polymers and their interaction with and effects on biological entities, polymer chemists need to work closely together with pharmacists, biologists and medical doctors.From the point of view of the authors, major improvements in the field of polymer based nanomedicine can be expected whenever advanced polymer chemistry is combined with biological rationale of the disease to be cured. However, as we are still not understanding the complex interactions of the plethora of synthetic materials with the variety of biological entities and barriers in the human organism many questions remain to be answered and maybe more to be asked with the final goal to improve the quality of life for many patients.
Acknowledgements
R.L. gratefully acknowledges support by the King Abdullah University of Science and Technology, KAUST (award no. KUK-F1-029–32) and ongoing support by Prof. R. Jordan. M.B. would like to thank the graduate school of excellence MAINZ, COMATT, IMPRS, the Spanish Ministry Grant (CTQ2007-606019), European Commission FP7-Health program (proposal n. 241919, LIVIMODE) and particularly the cluster of excellence SAMT of Rhineland-Palatina for support and funding.References
- H. Jatzkewitz, Hoppe-Seyler's Z. Physiol. Chem., 1954, 297, 149–156 Search PubMed.
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 Search PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- Y. Matsumura and K. Kataoka, Cancer Sci., 2009, 100, 572–579 CrossRef CAS.
- E. V. Batrakova and A. V. Kabanov, J. Controlled Release, 2008, 130, 98–106 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- A. Samad, Y. Sultana and M. Aqil, Curr. Drug Delivery, 2007, 4(4), 297–305 Search PubMed.
- D. A. LaVan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21(10), 1184–1191 CrossRef.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 Search PubMed.
- V. Mailänder and K. Landfester, Biomacromolecules, 2009, 10(9), 2379–2400 CrossRef.
- J. M. Harris and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55(10), 1259–1260 Search PubMed.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61(13), 1177–1188 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- M. A. Gauthier and H.-A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35(1), 9–16 Search PubMed.
- I. Hamad, A. C. Hunter, J. Szebeni and S. M. Moghimi, Mol. Immunol., 2008, 46, 225–232 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter, C. M. Dadswell, S. Savay, C. R. Alving and J. Szebeni, Biochim. Biophys. Acta, 2004, 1689, 103–113 Search PubMed.
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354, 56–62 CrossRef CAS.
- V. Kumar and D. S. Kalonia, Pharm. Sci. Technol., 2006, 7, 62 Search PubMed.
- H.-J. Sung, P. Chandra, M. D. Treiser, E. Liu, C. P. Iovine, P. V. Moghe and J. Kohn, J. Cell. Physiol., 2009, 218, 549–557 CrossRef CAS.
- H.-J. Sung, A. Luk, N. S. Murthy, E. Liu, M. Jois, A. Joy, J. Bushman, P. V. Moghe and J. Kohn, Soft Matter, 2010, 6, 5196–5205 RSC.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discov. Today, 2005, 10, 1451–1458 CrossRef CAS.
- M. K. Pratten, J. B. Lloyd, G. Hörpel and H. Ringsdorf, Makromol. Chem., 1985, 186, 725–733 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82(2–3), 189–212 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- Characterisation and Analysis of Polymers, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- P. C. Hiemenz, Polymer Chemistry, Marcel Decker INC, New York, Basel, 1984 Search PubMed.
- W. Buchard, Advances in Polymer Science, Springer-Verlag, Berlin, Heidelberg, 1983, p. 48 Search PubMed.
- H. J. A. Philipsen, J. Chromatogr., A, 2004, 1037(1–2), 329–350 CrossRef.
- G. Montaudo and M. S. Montaudo, Mass Spectrometry of Polymers, CRC Press LLC, Boca Raton, 2002 Search PubMed.
- G. Thews, E. Mutschler and P. Vaupel, Anatomie, Physiologie, Pathophysiologie des Menschen, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, 6th edn, 2007 Search PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS.
- D. C. Schriemer and L. Li, Anal. Chem., 1997, 69, 4169–4175 CrossRef.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- H. Pan, J. Yang, P. Kopečková and J. Kopeček, Biomacromolecules, 2011, 12, 247–252 Search PubMed.
- H. Gemici, T. M. Legge, M. Whittaker, M. J. Monteiro and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2334–2340 CrossRef CAS.
- M. H. Sung, C. Park, C. J. Kim, H. Poo, K. Soda and M. Ashiuchi, Chem. Rev., 2005, 5(6), 352–366 Search PubMed.
- C. Li, Adv. Drug Delivery Rev., 2002, 54(5), 695–713 CrossRef CAS.
- T. Candela and A. Fouet, Mol. Microbiol., 2006, 60, 1091–1098 CrossRef CAS.
- (a) H. Leuchs, Ber. Dtsch. Chem. Ges., 1906, 39, 857–861 CrossRef; (b) H. Leuchs and W. Manasse, Ber. Dtsch. Chem. Ges., 1907, 40, 3235–3249 CAS; (c) H. Leuchs and W. Geiger, Ber. Dtsch. Chem. Ges., 1908, 41, 1721–1726 Search PubMed.
- H. R. Kricheldorf, α-Aminoacid-N-Carboxyanhydrides and Related Materials, Springer, New York, 1987 Search PubMed.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2008, 130, 12562–12563 CrossRef CAS.
- M. Meyer and H. Schlaad, Macromolecules, 2006, 39, 3967–3970 CrossRef CAS.
- C. Scholz, W. Vayaboury, Int. Pat. Application, 2008, PCT/US2008/012258.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4670–4673 Search PubMed.
- T. Higashihara and R. Faust, React. Funct. Polym., 2009, 69, 429–434 Search PubMed.
- R. Nakamura, K. Aoi and M. Okada, Macromol. Rapid Commun., 2006, 27, 1725–1732 Search PubMed.
- T. Aliferis, H. Iatrou, N. Hadjichristidis, J. Messman and J. Mays, Macromol. Symp., 2006, 240, 12–17 Search PubMed.
- H. Lu, J. Wang, Y. Lin and J. Cheng, J. Am. Chem. Soc., 2009, 131, 13582–13583 CrossRef CAS.
- H. Yuan, K. Luo, Y. Lai, Y. Pu, B. He, G. Wang, Y. Wu and Z. Gu, Mol. Pharmacol., 2010,(7), 953–962 Search PubMed.
- A. Makino, S. Kizaka-Kondoh, R. Yamahara, I. Hara, T. Kanzaki, E. Ozeki, M. Hiraoka and S. Kimura, Biomaterials, 2009, 30, 5156–5160 CrossRef CAS.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2008, 19, 109–117 CrossRef CAS.
- T. Kanzaki, Y. Horikawa, A. Makino, J. Sugiyama and S. Kimura, Macromol. Biosci., 2008, 8, 1026–1033 CrossRef CAS.
- S. Kimura, T. Kidchob and Y. Imanishi, Polym. Adv. Technol., 2001, 12, 85–95 CrossRef CAS.
- B. Romberg, J. M. Metselaar, L. Baranyi, C. J. Snel, R. Bünger, W. E. Hennink, J. Szebeni and G. Storm, Int. J. Pharm., 2007, 331, 186–189 CrossRef CAS.
- K. Aoi, T. Hatanaka, K. Tsutsumiuchi, M. Okada and T. Imae, Macromol. Rapid Commun., 1999, 20, 378–382 Search PubMed.
- J. M. Metselaar, P. Bruin, L. W. de Boer, T. de Vringer, C. Snel, C. Oussoren, M. H. Wauben, D. J. Crommelin, G. Storm and W. E. Hennink, Bioconjugate Chem., 2003, 14, 1156–1164 CrossRef CAS.
- W. Tansey, S. Ke, X. Y. Cao, M. J. Pasuelo, S. Wallace and C. Li, J. Controlled Release, 2004, 94, 39–51 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- L. Guo and D. Zhang, J. Am. Chem. Soc., 2009, 131, 18072–18074 Search PubMed.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- M. Beck, P. Birnbrich, U. Eicken, H. Fischer, W. E. Fristad, B. Hase and H.-J. Krause, Angew. Makromol. Chem., 1994, 223, 217–233 CrossRef CAS.
- M. Litt, A. Levy and J. Herz, J. Polym. Sci., Polym. Chem. Ed., 1975, 9, 703–727 Search PubMed.
- O. Nuyken, G. Maier, A. Groß and H. Fischer, Macromol. Chem. Phys., 1996, 197, 83–95 Search PubMed.
- R. Luxenhofer and R. Jordan, Macromolecules, 2006, 39, 3509–3516 CrossRef CAS.
- S. Kobayashi, E. Masuda, S. Shoda and Y. Shimano, Macromolecules, 1989, 22, 2878–2884 CrossRef CAS.
- G. H. Hsiue, H. Z. Chiang, C. H. Wang and T. M. Juang, Bioconjugate Chem., 2006, 17, 781–786 CrossRef CAS.
- J. M. Harris, M. D. Bentley, K. Yoon, Z. Fang, F. M. Veronese, US Pat. Application, 2010, US2010/0069579.
- M. B. Foreman, J. P. Coffman, M. J. Murcia, S. Cesana, R. Jordan, G. S. Smith and C. A. Naumann, Langmuir, 2003, 19, 326–332 CrossRef CAS.
- M. Lobert, U. Köhn, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 1458–1460 RSC.
- C. Taubmann, R. Luxenhofer, S. Cesana and R. Jordan, Macromol. Biosci., 2005, 5, 603–612 CrossRef CAS.
- W. H. Binder and H. Gruber, Macromol. Chem. Phys., 2000, 201, 949–957 CrossRef CAS.
- A. Levy and M. Litt, J. Polym. Sci., Part A-1, 1968, 6, 1883–1894 Search PubMed.
- S. Cesana, A. Kurek, M. A. Baur, J. Auernheimer and O. Nuyken, Macromol. Rapid Commun., 2007, 28, 608–615 CrossRef CAS.
- S. Cesana, J. Auernheimer, R. Jordan, H. Kessler and O. Nuyken, Macromol. Chem. Phys., 2006, 207, 183–192 CrossRef CAS.
- J. Kronek, J. Luston, Z. Kroneková, E. Paulovicová, P. Farkas, N. Petrencíková, L. Paulovicová and I. Janigová, J. Mater. Sci.: Mater. Med., 2010, 21, 879–886 Search PubMed.
- M. Barz, R. Luxenhofer, R. Zentel and A. V. Kabanov, Biomaterials, 2009, 30, 5682–5690 Search PubMed.
- R. Luxenhofer, M. Bezen and R. Jordan, Macromol. Rapid Commun., 2008, 29, 1509–1513 Search PubMed.
- N. Zhang, A. Schulz, S. Huber, R. Luxenhofer and R. Jordan, Macromolecules, 2008, 42, 2215–2221 Search PubMed.
- N. Zhang, M. Steenackers, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 5345–5351 CrossRef CAS.
- S. Kobayashi, H. Uyama, Y. Narita and J. Ishiyama, Macromolecules, 1992, 25, 3232–3236 CrossRef CAS.
- S. C. Lee, C. Kim, I. C. Kwon, H. Chung and S. Y. Jeong, J. Controlled Release, 2003, 89, 437–446 CrossRef CAS.
- K. Tsutsumiuchi, K. Aoi and M. Okada, Macromolecules, 1997, 30, 4013–4017 CrossRef CAS.
- C.-H. Wang, C. H. Wang and G. H. Hsiue, J. Controlled Release, 2005, 108, 140–149 Search PubMed.
- C.-H. Wang and G.-H. Hsiue, Biomacromolecules, 2003, 4, 1537–1490 Search PubMed.
- C.-H. Wang, W.-T. Wang and G.-H. Hsiue, Biomaterials, 2009, 30, 3352–3358 CrossRef CAS.
- M. M. Bloksma, M. M. R. M. Hendrix, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 4654–4659 CrossRef CAS.
- M. M. Bloksma, S. Rogers, U. S. Schubert and R. Hoogenboom, Soft Matter, 2010, 6, 994–1003 RSC.
- M. M. Bloksma, U. S. Schubert and R. Hoogenboom, Polym. Chem., 2011, 2, 203–208 RSC.
- (a) S. Zalipsky, C. B. Hansen, J. M. Oaks and T. M. Allen, J. Pharm. Sci., 1996, 85, 133–137 CrossRef CAS; (b) M. C. Woodle, C. M. Engbers and S. Zalipsky, Bioconjugate Chem., 1994, 5, 493–496 CrossRef CAS.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS.
- O. Purrucker, A. Förtig, R. Jordan and M. Tanaka, ChemPhysChem, 2004, 5, 327–335 CrossRef CAS.
- O. Purrucker, S. Gönnenwein, A. Förtig, R. Jordan, M. Rusp, M. Bärmann, L. Moroder, E. Sackmann and M. Tanaka, Soft Matter, 2007, 3, 333–336 RSC.
- J. M. Harris, M. D. Bentley, T. X. Viegas, presented in part at the 8th International Symposium on Polymer Therapeutics: from Laboratory to Clinical Practice, Valencia, May, 2010.
- M. Miyamoto, K. Naka, M. Shiozaki, Y. Chujo and T. Saegusa, Macromolecules, 1990, 23, 3201–3205 Search PubMed.
- W. H. Velander, R. D. Madurawe, A. Subramanian, G. Kumar, G. Sinai-Zingde, J. S. Riffle and C. L. Orthner, Biotechnol. Bioeng., 1992, 39, 1024–1030 CrossRef.
- P. Farkas, J. Korcová, J. Kronek and S. Bystrický, Eur. J. Med. Chem., 2010, 45, 795–799 Search PubMed.
- R. Luxenhofer, M. López-García, A. Frank, H. Kessler and R. Jordan, PMSE Prepr., 2006, 95, 283–284 Search PubMed.
- R. Luxenhofer, PhD thesis, Technische Universität München, Garching, 2007.
- C. Diehl and H. Schlaad, Chem.–Eur. J., 2009, 15, 11469–11472 CrossRef CAS.
- F. Manzenrieder, R. Luxenhofer, M. Retzlaff, R. Jordan and M. G. Finn, Angew. Chem., Int. Ed., 2011, 50(11), 2601–2605 Search PubMed.
- K. Naka, A. Ohki and S. Maeda, Chem. Lett., 1991, 20, 1303–1306 Search PubMed.
- K. Naka, T. Nakamura, A. Ohki and S. Maeda, Polym. J. (Tokyo), 1995, 27, 1071–1078 Search PubMed.
- K. Naka, R. Yamashita, T. Nakamura, A. Ohki, S. Maeda, K. Aoi, K. Tsutsumiuchi and M. Okada, Macromol. Chem. Phys., 1997, 198, 89–100 CrossRef CAS.
- K. Naka, Y. Kubo, A. Ohki and S. Maeda, Polym. J. (Tokyo), 1994, 26, 243–249 Search PubMed.
- A. Ohki, K. Naka, O. Ito and S. Maeda, Chem. Lett., 1994, 23, 1065 Search PubMed.
- K. Naka, T. Nakamura, A. Ohki and S. Maeda, Macromol. Chem. Phys., 1997, 198, 101–116 Search PubMed.
- R. Konradi, B. Pidhatika, A. Mühlebach and M. Textor, Langmuir, 2008, 24, 613–616 CrossRef CAS.
- M. Grzelakowski, O. Onaca, P. Rigler, M. Kumar and W. Meier, Small, 2009, 5, 2545–2548 CrossRef CAS.
- A. Graff, C. Fraysse-Ailhas, C. G. Palivan, M. Grzelakowski, T. Friedrich, C. Vebert, G. Gescheidt and W. Meier, Macromol. Chem. Phys., 2009, 211, 229–238 Search PubMed.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS.
- H.-J. Choi and C. D. Montemagno, Nanotechnology, 2006, 17, 2198–2202 CrossRef CAS.
- H.-J. Choi, E. Brooks and C. D. Montemagno, Nanotechnology, 2005, 16, S143–S149 Search PubMed.
- H.-J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS.
- G. D. Bachand, R. K. Soong, H. P. Neves, A. Olkhovets, H. G. Craighead and C. D. Montemagno, Nano Lett., 2001, 1, 42–44 CrossRef CAS.
- S. Huber and R. Jordan, Colloid Polym. Sci., 2007, 286, 395–402 Search PubMed.
- J. S. Park and K. Kataoka, Macromolecules, 2007, 40, 3599–3609 CrossRef CAS.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653–1661 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS.
- (a) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS; (b) C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- K. Matyjaszewski, Y. Gnanou and L. Leibler, Macromolecular Engineering, Wiley-VCH, Weinheim, 2007, vol. 1 Search PubMed.
- F. J. Xu, K. G. Neoh and E. T. Kang, Prog. Polym. Sci., 2009, 34(8), 719–761 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60(9), 1018–1036 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34(4), 317–350 CrossRef CAS.
- H. R. Marsden and A. Kros, Macromol. Biosci., 2009, 9(10), 939–951 CrossRef CAS.
- M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel and J. Bernard, Macromol. Rapid Commun., 2010, 31(20), 1751–1772 CrossRef CAS.
- M. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, J. Mater. Chem. Rev., 2007, 107, 2270–2299 Search PubMed.
- J. H. Oh, S. A. Bencherif and K. Matyjaszewski, Polymer, 2009, 50, 4407–4423 CrossRef CAS.
- C.-W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- T. M. Legge, A. T. Slark and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6980–6987 CrossRef.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- E. A. Castro, Chem. Rev., 1999, 99, 3505–3524 CrossRef CAS.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446–7452 CrossRef CAS.
- M. F. Llauro, J. Loiseau, F. Boisson, F. Delolme, C. Ladaviere and J. Claverie, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5439–5462 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 26, 8312–8319 Search PubMed.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- L. Tao, G. Mantonani, F. Lecolley and D. M. Haddelton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- A. O. Saeed, S. Dey, S. M. Howdle, K. J. Thurecht and C. Alexander, J. Mater. Chem., 2009, 19, 4529–4535 RSC.
- L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123 CrossRef CAS.
- F. Ahmed, R. I. Paunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Mol. Pharmacol., 2006, 3, 340–350 CrossRef CAS.
- M. Eaton, Nat. Mater., 2007, 6, 251–253 Search PubMed.
- S. M. Ryan, X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51–59 CrossRef CAS.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(36), 15231–15236 CrossRef CAS.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- A. L. Lewis, Encycl. Biomater. Biomed. Eng., 2004, 1198–1211 Search PubMed.
- E. J. Lobb, I. Ma, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 2001, 123, 7913–7914 CrossRef CAS.
- Y. Ma, Y. Tang, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. P. Salvage, Macromolecules, 2003, 36, 3475–3484 CrossRef CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS.
- M. H. Stenzel, C. Barner-Kowollik, T. P. Davis and H. M. Dalton, Macromol. Biosci., 2004, 4, 445–453 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- M. Licciardi, Y. Tang, N. C. Billingham, S. P. Armes and A. L. Lewis, Biomacromolecules, 2005, 6, 1085–1098 CrossRef CAS.
- G. Battaglia and A. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS.
- V. Hearnden, H. Lomas, S. MacNeil, M. Thornhill, C. Murdoch, A. Lewis, J. Madsen, A. Blanazs, S. Armes and G. Battaglia, Pharm. Res., 2009, 26(7), 1718–1728 CrossRef CAS.
- H. Lomas, M. Massignani, K. A. Abdullah, I. Canton, C. L. Presti, S. MacNeil, J. Du, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Faraday Discuss., 2008, 139, 142–159 Search PubMed.
- A. Lewis, Y. Tang, S. Brocchini, J. Choi and A. Godwin, Bioconjugate Chem., 2008, 19(11), 2144–2155 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Polymer, 2003, 44, 6761–6765 CrossRef CAS.
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(22), 5031–5036 CrossRef CAS.
- S. G. Spain and N. R. Cameron, Polym. Chem., 2010, 1, 60–68 Search PubMed.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- A. Varki, R. D. Cummings, J. D. Esko, H. Freeze, G. Hart and J. Marth, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999 Search PubMed.
- Q. Yao and D. E. Furst, Rheumatology, 2008, 47, 946–951 Search PubMed.
- K. Baumann, D. Kowalczyk, T. Gutjahr, M. Pieczyk, C. Jones, M. K. Wild, D. Vestweber and H. Kunz, Angew. Chem., Int. Ed., 2009, 48, 3174–3178 Search PubMed.
- (a) D. Vestweber and J. E. Blanks, Physiol. Rev., 1999, 79, 181–213 CAS; (b) F. M. Unger, Adv. Carbohydr. Chem. Biochem., 2001, 57, 207–435 CAS; (c) R. P. McEver, Curr. Opin. Cell Biol., 2002, 14, 581–586 CrossRef CAS; (d) M. Sperandio, FEBS J., 2006, 273, 4377–4389 CrossRef CAS.
- Z. Deng, M. Ahmed and R. J. Narain, Polym. Sci., Part A: Polym. Chem., 2009, 47, 614–627 Search PubMed.
- A. M. Granville, D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Symp., 2007, 255, 81–89 CrossRef CAS.
- N. Ayres, D. J. Holt, C. F. Jones, L. E. Corum and D. W. Grainger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7713–7724 CrossRef CAS.
- A. Godwin, M. Hartenstein, A. H. E. Müller and S. Brocchini, Angew. Chem., Int. Ed., 2001, 40, 594–597 CrossRef CAS.
- C. W. Scales, F. Huang, N. Li, Y. A. Vasilieva, J. Ray, A. J. Convertine and C. L. McCormick, Macromolecules, 2006, 39, 6871–6881 CrossRef CAS.
- M. J. Yanjarappa, K. V. Gujraty, A. Joshi, A. Saraph and R. S. Kane, Biomacromolecules, 2006, 7, 1665–1670 CrossRef CAS.
- C.-Y. Hong and C.-Y. Pan, Macromolecules, 2006, 39, 3517–3524 CrossRef CAS.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- T. Lammers and K. Ulbrich, Adv. Drug Delivery Rev., 2010, 62(2), 119–121 Search PubMed.
- J. Kopeček and P. Kopečková, Adv. Drug Delivery Rev., 2010, 62(2), 122–149 Search PubMed.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2010, 62(2), 150–166 Search PubMed.
- B. Říhová and M. Kovář, Adv. Drug Delivery Rev., 2010, 62(2), 184–191 Search PubMed.
- T. Lammers, Adv. Drug Delivery Rev., 2010, 62(2), 203–230 Search PubMed.
- M. Talelli, C. J. F. Rijcken, C. F. van Nostrum, G. Storm and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62(2), 231–239 Search PubMed.
- Z.-R. Lu, Adv. Drug Delivery Rev., 2010, 62(2), 246–257 Search PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2010, 62(2), 272–282 Search PubMed.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 Search PubMed.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- R. Duncan, J. K. Coatsworth and S. Burtles, Hum. Exp. Toxicol., 1998, 17, 93–104 Search PubMed.
- J. W. Hopewell, R. Duncan, D. Wilding and K. Chakrabarti, Hum. Exp. Toxicol., 2001, 20, 461–470 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- M. Barz, M. Tarantola, K. Fischer, M. Schmidt, R. Luxenhofer, A. Janshoff, P. Theato and R. Zentel, Biomacromolecules, 2008, 9, 3114–3118 CrossRef CAS.
- M. I. Gibson, E. Fröhlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4332–4345 CrossRef CAS.
- M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rösch, Biomacromolecules, 2009, 10, 1697–1703 Search PubMed.
- E. Pedone, X. Li, N. Koseva, O. Alpar and S. Brocchini, J. Mater. Chem., 2003, 13, 2825–2837 RSC.
- J. Khandare, A. Mohr, M. Calderon, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31(15), 4268–4277 CrossRef CAS.
- M. Marsh and H. T. McMahon, Science, 1999, 285, 215–220 CrossRef CAS.
- A. Sorkin and M. von Zastrow, Nat. Rev. Mol. Cell Biol., 2009, 10, 609–622 Search PubMed.
- L. Rajendran, H.-J. Knölker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29–42 CrossRef CAS.
- G. Sahay, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2008, 19, 2023–2029 Search PubMed.
- G. Sahay, J. O. Kim, A. V. Kabanov and T. K. Bronich, Biomaterials, 2009, 31, 923–933 Search PubMed.
- G. Sahay, V. Gautam, R. Luxenhofer and A. V. Kabanov, Biomaterials, 2009, 31, 1757–1764 Search PubMed.
- Y. Nemoto, A. Borovkov, Y.-M. Zhou, Y. Takewa, E. Tatsumi and Y. Nakayama, Bioconjugate Chem., 2009, 20, 2293–2299 Search PubMed.
- J. Callahan, P. Kopeckova and J. Kopecek, Biomacromolecules, 2009, 10, 1704–1714 Search PubMed.
- S. W. C. Richardson, K.-L. Wallom, E. L. Ferguson, S. P. E. Deacon, M. W. Davies, A. J. Powell, R. C. Piper and R. Duncan, J. Controlled Release, 2008, 127, 1–11 Search PubMed.
- T. Lammers, W. E. Hennink and G. Storm, Br. J. Cancer, 2008, 99, 392–397 Search PubMed.
- J. Sanchis, F. Canal, R. Lucas and M. J. Vicent, Nanomedicine, 2010, 5(6), 915–935 Search PubMed.
- G. M. Springett, C. Takimoto, M. McNamara, J. H. Doroshow, S. Syed, E. Eastham, D. Spriggs, S. Pezzulli, G. Michelson and J. Dupont, J. Clin. Oncol., 2004, 22(14), 3127 Search PubMed.
- J. W. Singer, R. Bhatt, J. Tulinsky, K. R. Buhler, E. Heasley, P. Klein and P. de Vries, J. Controlled Release, 2001, 74, 243–247 CrossRef CAS.
- S. D. Chipman, F. B. Oldham, G. Pezzoni and J. W. Singer, Int. J. Nanomed., 2006, 1, 375–383 Search PubMed.
- L. Paz-Ares, H. Ross, M. O'Brien, A. Riviere, U. Gatzemeier, J. Von Pawel, E. Kaukel, L. Freitag, W. Digel, H. Bischoff, R. García-Campelo, N. Iannotti, P. Reiterer, I. Bover, J. Prendiville, A. J. Eisenfeld, F. B. Oldham, B. Bandstra, J. W. Singer and P. Bonomi, Br. J. Cancer, 2008, 98, 1608–1613 Search PubMed.
- K. Albain, C. Belani, P. Bonomi, K. J. O'Byrne, J. H. Schiller and M. Socinski, Clin. Lung Cancer, 2006, 7(6), 417–419 Search PubMed.
- N. U. Lin, L. M. Parker, S. E. Come, H. J. Burstein, M. Haldoupis, N. Ryabin, R. Gelman, E. P. Winer and L. N. Shulman, Invest. New Drugs, 2007, 25(4), 369–375 Search PubMed.
- M. J. Shieh, C. L. Peng, W. L. Chiang, C. H. Wang, C. Y. Hsu, S. J. Wang and P. S. Lai, Mol. Pharmaceutics, 2010, 7, 1244–1253 Search PubMed.
- R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich, E. V. Batrakova, R. Jordan and A. V. Kabanov, Biomaterials, 2010, 31, 4972–4979 Search PubMed.
- A. V. Kabanov, R. Jordan, R. Luxenhofer, Int. Pat. Application, 2009, PCT/EP2009/004655.
- A. Mero, G. Pasut, L. Dalla Via, M. W. M. Fijten, U. S. Schubert, R. Hoogenboom and F. M. Veronese, J. Controlled Release, 2008, 125, 87–95 CrossRef CAS.
- J. Tong, R. Luxenhofer, X. Yi, R. Jordan and A. V. Kabanov, Mol. Pharmaceutics, 2010, 7, 984–992 Search PubMed.
- E. Segal, H. Pan, P. Ofek, T. Udagawa, P. Kopeckova, J. Kopecek and R. Satchi-Fainaro, PLoS One, 2009, 4(4), e5233 Search PubMed.
- Y. Qiao, Y. Huang, C. Qiu, X. Yue, L. Deng, Y. Wan, J. Xing, C. Zhang, S. Yuan, A. Dong and J. Xu, Biomaterials, 2010, 31, 115–123 Search PubMed.
- S. Kirkland-York, Y. Zhang, A. E. Smith, A. W. York, F. Huang and C. L. McCormick, Biomacromolecules, 2010, 11(4), 1052–1059 CrossRef CAS.
- S. Üzgün, Ö. Akdemir, G. Hasenpusch, C. Maucksch, M. M. Golas, B. Sander, H. Stark, R. Imker, J.-F. Lutz and C. Rudolph, Biomacromolecules, 2010, 11, 39–50 CrossRef CAS.
- A. Fire, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS.
- M. I. Siomi, Adv. Drug Delivery Rev., 2009, 61, 668–671 Search PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS.
- W. Gao, Z. Xiao, A. Radovic-Moreno, J. Shi, R. Langer and O. C. Farokhzad, Methods Mol. Biol., 2010, 629, 53–67 Search PubMed.
- Y. Takakura, Adv. Drug Delivery Rev., 2010, 61(9), 667 Search PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS.
- M. Hemmelmann, C. Knoth, U. Schmitt, M. Allmeroth, D. Moderegger, M. Barz, K. Koynov, C. Hiemke, F. Rösch and R. Zentel, Macromol. Rapid Commun. DOI:10.1002/marc.201000810.
Footnote |
| † Both authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2011 |