Gas-induced solid state transformation of an organic lattice: from nonporous to nanoporous†
Jian
Tian
a,
Praveen
Thallapally
*a,
Jun
Liu
b,
Gregory J.
Exarhos
b and
Jerry L.
Atwood
*c
aEnergy and Environment Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, United States. E-mail: Praveen.Thallapally@pnl.gov; Fax: +1-509-371-7249
bFundamental and Computational Science Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, United States
cDepartment of Chemistry, University of Missouri-Columbia, Columbia, MO 65211, United States. E-mail: atwoodj@missouri.edu; Fax: +1-573-882-2754
First published on 16th November 2010
Abstract
A well-known organic host, tris-o-phenylenedioxycyclotriphosphazene, exists in two polymorphic guest-free forms: the thermodynamic nonporous high-density phase and the kinetic nanoporous low-density phase. Simple pressurization of the high density phase with CO2 brings a solid-state transformation to the low density phase, resulting in significant expansion of the crystal volume by 23%.
The physical properties of organic molecular materials and pharmaceuticals are strongly dependent on the arrangement of molecules in the solid state.1 Manipulation and control of the molecular packing is of great importance for the desired properties and functions of materials. In general, molecular materials are stabilized by weak intermolecular interactions, and therefore the solid state packing can be perturbed easily by external stimuli such as pressure, temperature and pH during which molecular materials may undergo structural transformation.2 In the present context, if the molecular material is a host, an increase in temperature leads to the removal of guest solvent molecules. This process is often reversible and generally results in the concomitant rearrangement of the solid state packing. As a result the original network is not retained, and is only re-obtained by the re-absorption of the same (or in some cases different) solvent molecules.3 In this regard, there are very few reports on ‘gas-driven’ phase transformations involving thermodynamically stable phases converting to kinetic phases.4 Herein, we report the gas-triggered transformation of a well-known organic host, tris-o-phenylenedioxycyclotriphosphazene (TPP, 1, Scheme 1), from its thermodynamic nonporous form to the kinetic porous form with hexagonal channels, as well as concomitant conformational change of host molecules.
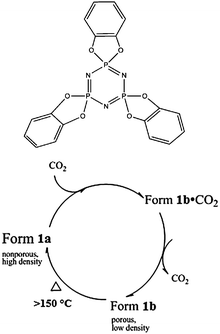 | ||
| Scheme 1 Schematic diagram of TPP (1) and its conversions from nonporous to porous form. | ||
TPP is the simplest member of a family of spirocyclic phosphazene molecules, discovered by Allcock and co-workers.5 Studied extensively as host materials, TPP is known to include a wide range of guest molecules, such as water, benzene, THF and I2.6,7 It also has shown promise in the area of gas storage and intriguing electro optical properties due to the extended π conjugated systems.7c–f Within the literature on the basis of this well studied host, it is known that thermodynamic (1a) and kinetic (1b) guest-free forms of 1 can respectively obtained by sublimation under vacuum or by careful desolvation of TPP benzene channel inclusion compound (1b·benzne).6,7d Crystals of pure guest-free 1a were prepared by triple sublimation at 175 °C under vacuum.6b Molecule 1 in 1a resembles a three-bladed paddle wheel in which the spirocyclic side groups are perpendicular to the plane of the central phosphazene ring (see ESI, Fig. S2†). It is noted that the side groups are bent at the bridging oxygen atoms away from the perpendicular symmetry plane positions of the central ring. As a result, the molecule is bent away from the trigonal symmetry and has concomitantly low Cs symmetry.8 Each molecule (molecular volume ∼321 Å3) occupies ∼468 Å3 of space in the monoclinic lattice, calculated from the unit cell parameters.‡
Crystals of guest-free 1b were obtained by careful evacuation of solvent molecules in TPP·benzene, as reported by Sozzani and co-workers.7d The host molecules in 1b have D3h symmetry and form a hexagonal array of microporous channels about 5 Å in diameter. With a large empty pore in the structure, each molecule of 1 occupies ∼577 Å3 of lattice volume, representing a 23% increase compared to the monoclinic system. The packing efficiency (PE) of 1b is 0.56 and the value is significantly lower than 1a (0.69), among the lowest in the molecular organic crystals.9 As a result, 1b has a low density (1.321 g cm−3vs. 1.628 g cm−3, 1a) with 25% of the volume is available to trap guest molecules in the noncovalent architecture.
It is well known that exposure of form 1a to solvent vapors brings a transition to hexagonal inclusion complex, with the solvent molecules incorporated into the nanochannels.6a However, guest-free hexagonal crystals (1b) are difficult to obtain by simple evacuation of solvent molecules (except for 1b·benzne). Complete removal of the included guest molecules generally causes the collapse of the hexagonal host lattice to the more efficiently packed monoclinic system.6 Previously, we and others have reported the unexpected solid-state transformation of guest-free p-tert-butylcalix[4]arene (TBC4) induced by gas.4b–d The nonporous, high-density form of TBC4 crystals could be expanded and transformed to porous, low-density forms when pressurized by CO2 or N2O. With this in mind, it is very interesting to investigate the gas effect on TPP guest-free forms, which also exist as a nonporous, high density form (1a) and a porous, low density form (1b) (Fig. 1).
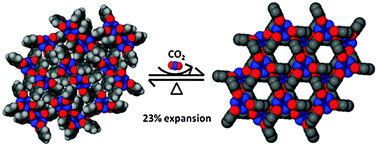 | ||
| Fig. 1 Gas-induced conversion of a nonporous high density phase 1a (left) to hexagonal porous phase 1b (right) with moderate pressures of CO2. | ||
The effects of various gas (CO2, N2O, CH4, He, and H2) pressures on low and high density polymorphic forms of 1 were examined by using in situ high pressure powder X-ray diffraction (PXRD) experiments. PXRD patterns of 1b with various gas pressures confirm no structural change, suggesting that 1b is stable with trapped guest gas molecules in the hexagonal nanochannels. However, when the high density form 1a was pressurized with 350 psi of CO2 (23.8 atm), the low density form 1b began to emerge as confirmed by the PXRD. The transformation completed within 3 h and resulted in a pure 1b phase (Fig. 2). The same conversion took place at even lower pressure of CO2 (150 psi), but over a longer time frame (2 days).
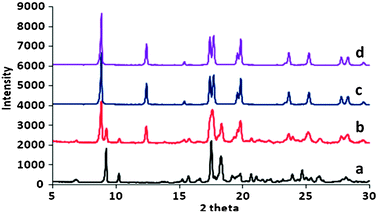 | ||
| Fig. 2 In situ conversion of 1a to 1b at 350 psi of CO2 at 298 K. (a) Nonporous form, 1a, (b) 1a transformed to 1b after exposing to 350 psi of CO2 for 1 h, (c) for 3 h and (d) after removal of CO2 under vacuum. | ||
To further support our evidence of transformation, the high density form 1a was loaded in a custom built volumetric gas analyzer and pressurized with 350 psi of CO2 gas. The pressure drop was monitored as a function of time. As shown in Fig. 3, the pressure decreases in the sample holder containing 1a, implying the gas molecules diffuse through the nonporous solid, converting the solid to porous form 1b. At the equilibrium point, the total uptake of CO2 by the solid is ∼12 wt% (60 cm3/g at STP) that is consistent with the reported value of 1b by Sozzani and co-workers, implying gas molecules were included in the hexagonal channels and in a short contact with the aromatic walls of the host framework.7d It is noted that the stored CO2 molecules would escape from the adsorption sites on release of gas pressure and leave a stable empty-channel 1b structure. As a consequence, the whole volume of 1a was significantly expanded by 23%. The whole transition can be best described as a two-step process by the following equations:
| 1a(solid) + CO2(gas) → 1b·CO2 + ΔEadsorption | (1) |
| 1b·CO2 → 1b(solid) + CO2(gas) + ΔEdesorption | (2) |
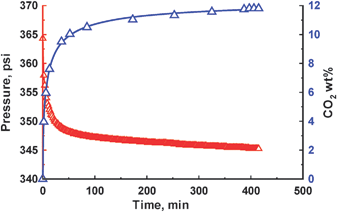 | ||
| Fig. 3 Conversion of 1a to 1b as function of gas loading and time at 298 K. | ||
In previous studies we have discussed the mechanism for gas-induced solid-state transformation of nonporous TBC4. Although TBC4 molecules pack closely within crystals, there remain lattice voids of ∼40 Å3 between layers of host molecules.4b These voids were believed to be the key factors for accommodating CO2 molecules and thus triggering the transformation. As for 1a, calculations based on packing of host molecules reveal that very small voids with a volume less than a hydrogen atom exist within the lattice.10 Such voids would not be large enough to host any guest molecules. As this is the case, the presence of large lattice voids suitable to accommodate guest gas molecules is not a requirement for effective gas-induced solid-state transformation. It is more likely that gas molecules first initiate the transition at the surface of 1a crystals by fast collision with the surface host molecules. We propose two possibilities: (1) As a response to collision, molecules of 1 at the crystal surface may change their conformation from the Cs symmetry to D3h symmetry, allowing the molecules to achieve less efficient packing, and thus create a transition zone with lattice voids/hexagonal channels that can accommodate guest molecules; (2) Surface molecules of 1a on specific crystal faces may already be in the open channel mode served as transition zones. In both cases, the tunnels formed at the transition interface would serve as conduits for the penetration of additional gases molecules and then propagate through the monoclinic lattice when the collision stimuli continues, affording the hexagonal lattice. Considering that a highly crystalline phase was obtained as the ‘product’, extensive cooperativity must exist between host molecules throughout the crystals during the transition.11
To conclude, we have demonstrated a gas-induced phase transformation of a well known host TPP from a high density nonporous form to a low density porous form with a 23% unit cell volume expansion by conformational changing from Cs to D3h symmetry. Such an expedient approach may hold great implications for the polymorphic control of molecular materials and pharmaceutical solids, a feature we are currently investigating. Our discovery also provides new opportunities for the study of solid state physics underlying such gas-induced transformations.
We acknowledge US Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award KC020105-FWP12152. PNNL is a multiprogram national laboratory operated for DOE by Battelle under Contract DE-AC05-76RL01830.
Notes and references
- W. Jones, Organic Molecular Solids: Properties and Applications, CRC Press, New York, 1997 Search PubMed.
- (a) K. Uemura, S. Kitagawa, K. Fukui and K. Saito, J. Am. Chem. Soc., 2004, 126, 3817 CrossRef CAS; (b) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS.
- (a) L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631 CrossRef CAS; (b) J. A. Ripmeester, G. D. Enright, C. I. Ratcliffe, K. A. Udachin and I. L. Moudrakovski, Chem. Commun., 2006, 4986 RSC.
- (a) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS; (b) P. K. Thallapally, P. B. McGrail, S. J. Dalgarno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146 CrossRef CAS; (c) P. K. Thallapally, P. B. McGrail, S. J. Dalgarno and J. L. Atwood, Cryst. Growth Des., 2008, 8, 2090 CrossRef CAS; (d) K. A. Udachin, I. L. Moudrakovski, G. D. Enright, C. I. Ratcliffe and J. A. Ripmeester, Phys. Chem. Chem. Phys., 2008, 10, 4636 RSC.
- (a) H. R. Allcock, J. Am. Chem. Soc., 1964, 86, 2591 CrossRef CAS; (b) H. R. Allcock and L. A. Siegel, J. Am. Chem. Soc., 1964, 86, 5140 CrossRef CAS.
- (a) H. R. Allcock, Acc. Chem. Res., 1978, 11, 81 CrossRef CAS; (b) H. R. Allcock, M. L. Levin and R. R. Whittle, Inorg. Chem., 1986, 25, 41 CrossRef CAS.
- (a) P. Sozzani, A. Comotti, R. Simonutti, T. Meersmann, J. W. Logan and A. Pines, Angew. Chem., Int. Ed., 2000, 39, 2695 CrossRef CAS; (b) T. Hertzsch, F. Budde, E. Weber and J. Hulliger, Angew. Chem., Int. Ed., 2002, 41, 2281 CrossRef CAS; (c) P. Sozzani, A. Comotti, S. Bracco and R. Simonutti, Angew. Chem., Int. Ed., 2004, 43, 2792 CrossRef CAS; (d) P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816 CrossRef CAS; (e) G. Couderc, T. Hertzsch, N.-R. Behrnd, K. Krämer and J. Hulliger, Microporous Mesoporous Mater., 2006, 88, 170 CrossRef CAS; (f) T. Hertzsch, C. Gervais, J. Hulliger, B. Jaeckel, S. Guentay, H. Bruchertseifer and A. Neels, Adv. Funct. Mater., 2006, 16, 268 CrossRef CAS; (g) A. Comotti, S. Bracco, L. Ferretti, M. Mauri, R. Simonutti and P. Sozzani, Chem. Commun., 2007, 350 RSC.
- Note: The molecular conformations of TPP in both 1a and 1b are approximate. In 1a, the molecular conformation is averaged for two unique 1 molecules in the asymmetric unit and has a noteworthy deviation from ideal Cs symmetry.
- (a) L. J. Barbour, Chem. Commun., 2006, 1163 RSC; (b) S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236 RSC.
- Computer program MSROLL was used for calculations with a probe radius 1.17 Å. M. L. Connolly, Science, 1983, 221, 709 Search PubMed.
- J. Tian, P. K. Thallapally, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2009, 131, 13216 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, Structural description of 1a and 1b, PXRD, and 13C solid state NMR study of transformation. See DOI: 10.1039/c0cc04260a |
| ‡ Crystallographic parameters of 1a and 1b: The expansion of crystal volume by 23% due to the solid state transformation was calculated from the reported single crystal structures of 1a and 1b. 1a (CSDDOFSUM):6bC36H24N6O12P6, M = 918.43, monoclinic, space groupP21/n, a = 24.969(6), b = 5.8268(14), c = 25.895(6) Å, β = 96.015(3)°, V = 3746.7(15) Å3, Z = 4; 1b(CSD DOFSUM01):7dC3H2N0.5OP0.5, M = 76.54, hexagonal, space groupP63/m, a = b = 11.454, c = 10.16 Å, α = β = 90°, γ = 120°, V = 1154.35 Å3, Z = 12. |
| This journal is © The Royal Society of Chemistry 2011 |