Zipper assembly of SHJ photosystems: focus on red naphthalenediimides, optoelectronic finetuning and topological matching†
Rajesh
Bhosale
a,
Ravuri S. K.
Kishore
a,
Velayutham
Ravikumar
a,
Oksana
Kel
b,
Eric
Vauthey
*b,
Naomi
Sakai
*a and
Stefan
Matile
*a
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Fax: +41 22 379 5123; Tel: +41 22 379 6523 Web: http://www.unige.ch/sciences/chiorg/matile/
bDepartment of Physical Chemistry, University of Geneva, Geneva, Switzerland
First published on 23rd June 2010
Abstract
The objective of this study was to synthesize multichromophoric donor–acceptor systems with non-halogenated red (RO) naphthalenediimides (NDIs) attached along p-oligophenyl (POP) and oligophenylethynyl (OPE) scaffolds, and to evaluate their usefulness for zipper assembly of artificial photosystems. Compared to halogenated red NDIs (RCl, RBr), the HOMO of RO is 0.2 eV higher and the HOMO/LUMO gap 0.1 eV smaller, the latter introducing a shade of pink. Consistent with higher HOMO levels, RO zippers generate less photocurrent than RBr zippers in their respective action spectra. RO zippers are less sensitive to topological mismatch than RBr zippers and thus more robust and broadly applicable. Transient absorption measurements reveal efficient electron transfer from excited OPE donors to RO acceptors and less efficient hole injection from excited RO donors into OPE acceptors. Both processes demonstrate compatibility with OMARG-SHJ photosystems (supramolecular n/p-heterojunctions with oriented multicolored antiparallel redox gradients). Decreasing hole transfer with decreasing HOMO energy differences further demonstrates that SHJ-type hole injection disappears gradually (rather than abruptly). Losses in photonic energy during this process can thus be minimized by optoelectronic finetuning, but eventual gains in open circuit voltages risk coming with complementary losses in short circuit current.
Introduction
In bilayer organic solar cells, exciton dissociation at the interface is followed by electron and hole translocation in n- and p-semiconducting bulk layers.1,2 In bulk n/p-heterojunction (BHJ) solar cells, maximized active interfaces produce more electrons (e−) and holes (h+) that are, however, less mobile.3–9 Increasing organization of BHJs leads to the situation where continuous e− and h+ transporting channels are aligned co-axially on the molecular level, an architecture that has been referred to as a supramolecular n/p-heterojunction (SHJ).1,10,11 Lessons from nature call for SHJs that are multicolored to absorb as much light as possible and contain an oriented antiparallel redox gradient (i.e., OMARG-SHJs) to quickly and safely move electrons and holes in opposite directions toward their final destinations.1 The construction of such OMARG-SHJs1 is far beyond reach despite inspired contributions from many groups. To build oriented SHJ architectures or redox cascades on solid substrates, surface-initiated polymerization of polymer brushes,12 surface-initiated electropolymerization,13,14 surface-initiated supramolecular polymerization,15,16 ordered17–19 or multicomponent20–22 versions of layer-by-layer (LBL) assembly17–21 or vapor deposition22 as well as zipper assembly23–27 have been considered.1 Zipper assembly has been introduced to construct e− transporting π-stacks along h+ transporting strings of rod-shaped oligomers. Naphthalenediimides (NDIs)28–34 as stacks and p-oligophenyls (POPs) and oligophenylethynyls (OPEs)7,35–40 as rods have been studied for zipper assembly because their optoelectronic properties are ideal for the construction of OMARG-SHJs (Fig. 1 and 2).23–27 For this purpose, multichromophoric POP-NDI hybrids 1–10 have been synthesized as propagators for zipper assembly with POP-NDI initiator 11 and LBL assembly with lipoic acid 12 (Fig. 1 and 3).23–26 In the more recent OPE series, OPE-NDI hybrids 13–16 have been realized as propagators for zipper assembly with OPE-NDI initiator 17 (Fig. 2 and 3).27,41,42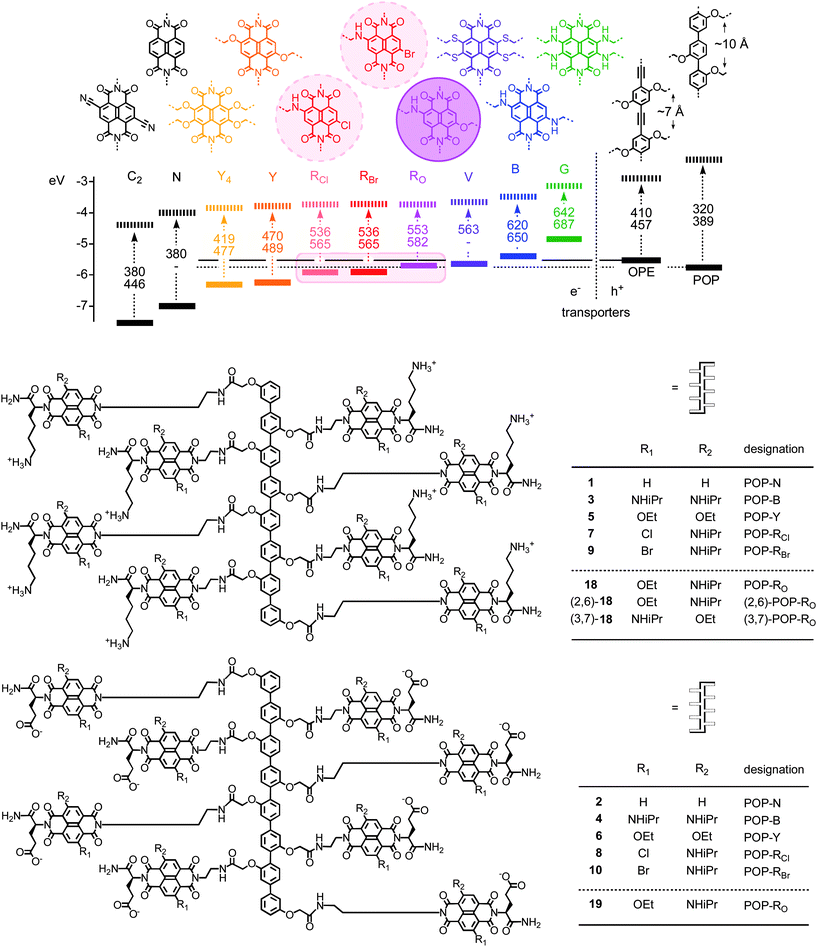 | ||
| Fig. 1 Frontier orbital energy levels of NDIs (general structures C2-G), POPs and OPEs (solid lines, HOMO; dashed lines, LUMO; dashed arrows, absorption of light (hν); with wavelength (nm) of maximal absorption (top) and emission (bottom), data from ref. 23–27,29,32, and this report) and structure of multicolor POP-NDI propagators 1–10 (previous)23–27,42 and 18–19 (this report). Unless indicated by numbers in parenthesis (e.g. (2,6)-18), all NDIs with two different substituents in the core and two different substituents in the periphery are mixtures of 2,6- and 3,7-regioisomers. This includes POP-NDIs 7–10, 18 and 19. RO is used as general designation for red NDIs with one alkoxy and one alkylamino substituent, RBr for red NDIs with one bromo and one alkylamino substituent, etc. | ||
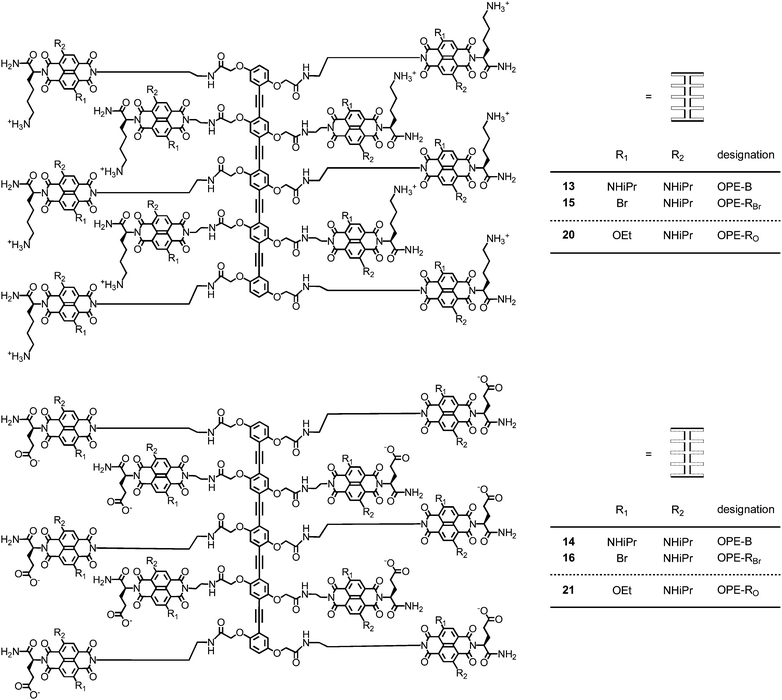 | ||
| Fig. 2 Structure of multicolor OPE-NDI propagators 13–16 (previous)27,42 and 20–21 (this report). OPE-NDIs 15, 16, 20 and 21 are mixtures of 2,6- and 3,7-regioisomers. | ||
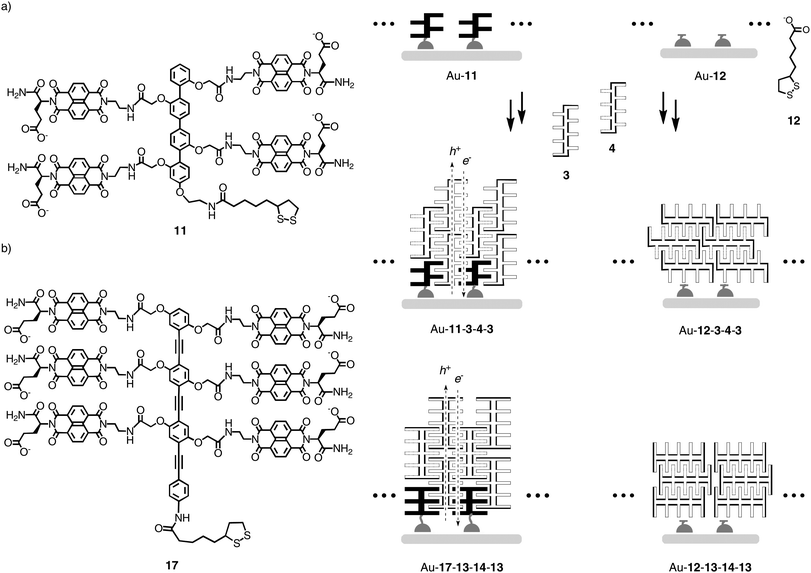 | ||
| Fig. 3 (a) Zipper assembly and LBL assembly of POP-NDI architectures on gold substrates, see Fig. 1 and 2 for structures of 1–16 (all suprastructures are speculative representations that are consistent with experimental data and drawn with the only intention to clarify the described concepts). (b) Analogous zipper and LBL architectures of OPE-NDIs. | ||
POP initiators 11 contain a disulfide at one end to covalently bind to the gold surface and negatively charged NDI acceptors (N) along the rigid-rod scaffold.23–27 Zipper assembly is then accomplished by dipping of Au-11 monolayers into solutions of POP-NDI hybrids of double length and complementary charge, such as the blue POP-B propagator 3, the yellow POP-Y propagator 5 or the red POP-RBr propagator 9 (Fig. 3a). The lower half of, e.g., the blue NDIs of propagator 3, should interdigitate with the NDIs of initiator 11 to form π-stacks that are organized by intrastack hydrogen bonds and interstack ion pairs. The upper half of the blue NDIs of propagator 3 remains free as “sticky ends” at the surface of Au-11-3 to zip up with the anionic propagator 4, whose sticky ends at the surface of Au-11-3-4 can zip up with the cationic propagator 3, and so on.
The obtained zipper architecture Au-11-(3-4-)n is composed of π-stacks formed by interdigitating aromatic planes from neighboring scaffolds along strings of interdigitating rigid rods. The results are operational inter- and intralayer recognition motifs that direct the co-axial alignment of electron transporting NDI stacks next to hole transporting POP rods. Zipper architectures have been routinely characterized in comparison with the complementary LBL assembly, where initiators 11 or 17 are replaced by lipoic acid 12 (Fig. 3). The more disordered LBL architectures reproducibly form faster, have rougher surfaces and reduced critical thickness, and generate less photocurrent than the more ordered zipper architectures.26,27,42
OPE scaffolds in zipper architectures have been introduced as attractive partners for POPs in zipper assembly because their high HOMO is compatible with the construction of redox gradients in the hole conducting channel (Fig. 1).27 Moreover, the repeat distance of ∼7 Å between side chains attached along the OPE scaffold perfectly matches the repeat distance of π-stacks formed by interdigitating arene planes that are separated by ∼3.5 Å (Fig. 1). POP scaffolds have a repeat of 10 Å, which is too much to preorganize co-axial π-stacks with high precision.43 The situation with OPEs has been referred to as rod/stack (or topological) matching, and that with POPs as rod/stack mismatch.
The influence of topological matching in the molecular building blocks on the organization of the final supramolecular system can vary substantially. Supramolecular architectures that are disorganized by mismatched building blocks are referred to as mismatch sensitive, those resisting topological mismatch as mismatch insensitive. POP zipper architectures with brominated NDI chromophores RBr are mismatch sensitive, whereas yellow (Y) or blue (B) NDIs tolerate mismatched building blocks better.26,27,42 The origin of the mismatch sensitivity of RBr is unknown, excessive hydrophobicity, missing dispersion contacts between stacks or even competing halogen bonds44,45 deserve consideration.
Another weakness of RBr photosystems concerns their optoelectronic properties. The difference in HOMO energies between RBr hole donors and particularly OPE hole acceptors suggests that photoinduced charge separation (PCS) occurs with a considerable loss in photonic energy. Moreover, PCS and charge recombination (CR) is complicated with halogenated NDIs because of triplet contributions from heavy atom effects.27
The objective of this study was to explore the ability of non-halogenated but equally red OPE-RO and POP-RO zippers to overcome the sensitivity of brominated red zippers toward topological mismatch on the one hand and to minimize energy losses during rod/stack PCS on the other. RO fluorophores, i.e., NDIs with one alkoxy and one alkylamino substituent in the core, have been introduced early on.46 However, RO fluorophores have never been used before for zipper assembly of SHJ photosystems. The HOMO energy of RO fluorophores is higher than that of RBr and RCl (Fig. 1). This reduces the energy gap to POP and OPE hole acceptors. The nearly identical HOMO levels of RO donors and OPE acceptors suggested that SHJ-type rod/stack PCS, if possible, would occur with minimal losses in photonic energy. In the following, we report synthesis and evaluation of OPE-RO and POP-RO photosystems and show that they fully live up to the above expectations.
Results and discussion
Synthesis
The synthesis of the RO chromophore via Cl, alkoxy substituted NDI has been previously reported by Würthner and coworkers.46 We have found that this chromophore can be more conveniently prepared by the reaction of diethoxysubstituted NDIs with alkylamines.47 The cationic and anionic POP-RO hybrids 18 and 19 were synthesized from naphthalenedianhydride 22 (Schemes 1 and 2). The transformation of the colorless dianhydride 22 into the yellow NDI 23 was accomplished following previously reported procedures.26,47 Nucleophilic aromatic core substitution with isopropylamine 24 under mild conditions gave the desired red RO chromophore 25. It could be obtained in high yield because the reduced π-acidity of the product prevented continuing nucleophilic aromatic substitution to the blue diisopropylamino NDI. The two regioisomers of 25 could be separated by PTLC. Running three-times in CH2Cl2–MeOH 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 as mobile phase, two overlapping bands in different shades of magenta were obtained around Rf 0.60 (Scheme 1). The less polar regioisomer was identified by HMBC spectra to be (3,7)-25. Some significant H to C crosspeaks are indicated with arrows in Scheme 1. Alloc removal with Pd(PPh3)4 and Bu3SiH proceeded without damage of the Z protecting group. The obtained amines 26 were used without workup to prevent cyclization into the amidines.
:3 as mobile phase, two overlapping bands in different shades of magenta were obtained around Rf 0.60 (Scheme 1). The less polar regioisomer was identified by HMBC spectra to be (3,7)-25. Some significant H to C crosspeaks are indicated with arrows in Scheme 1. Alloc removal with Pd(PPh3)4 and Bu3SiH proceeded without damage of the Z protecting group. The obtained amines 26 were used without workup to prevent cyclization into the amidines.
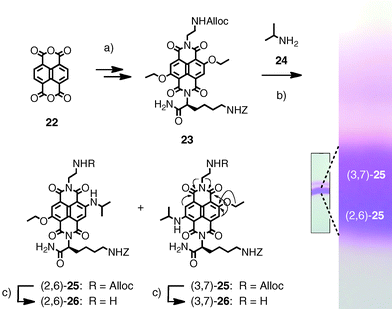 | ||
| Scheme 1 (a) Several steps.26 (b) 24, rt, 4 h, 77%. (c) Pd(PPh3)4, Bu3SiH, useable without work-up. Arrows in (3,7)-25 show significant H → C HMBC crosspeaks. | ||
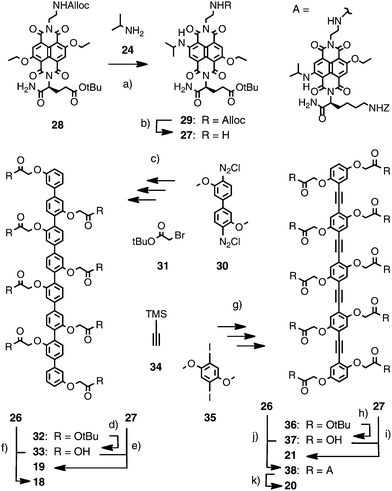 | ||
| Scheme 2 (a) 24, rt, 4 h, 42%. (b) Pd(PPh3)4, Bu3SiH, useable without work-up only. (c) several steps.48 (d) TFA.48 (e) 1. HATU, DTBP, DMF, rt, 16 h, 53%. 2. TFA, pentamethylbenzene, thioanisole, HBr, rt, 4 h, quant. (f) 1. HATU, DTBP, TEA, DMF, rt, 16 h, 52%. 2. TFA, rt, 1 h, quant. (g) several steps.27 (h) TFA.27 (i) 1. HATU, DTBP, DMF, rt, 16 h, 28%. 2. TFA, rt, 3 h, quant. (j) HATU, DTBP, TEA, DMF, rt, 16 h, 43%. (k) TFA, pentamethylbenzene, thioanisole, 50 °C, 3 h, quant. | ||
The corresponding amine 27 for the anionic POP-RO19 was prepared analogously from the yellow 28 by core substitution with amine 24 followed by chemoselective deprotection of the red chromophore 29 (Scheme 2). Contrary to the situation with cationic RO25, it was not possible to separate the two regioisomers of the anionic RO29.
The synthesis of POP scaffolds from biphenyl 30 and acetate 31 was first reported 11 years ago and has been extensively used and refined since then (Scheme 2).48 Deprotection of the key intermediate 32 with TFA gave POP 33 with acids lining the rigid-rod scaffold for coupling with amine 26. Amide bond formation followed by deprotection of the peripheral amines gave the cationic POP-NDI 18. Reaction of POP acid 33 with amine 27 followed by deprotection of the acids gave the anionic POP-NDI 19. The pure regioisomers (2,6)-26 and (3,7)-26 were used to prepare regiopure multichromophoric POP-RO systems (2,6)-18 and (3,7)-18. Comparison of regiopure (2,6)-18 and (3,7)-18 with the statistical mixture 18 did overall not reveal significant differences in photocurrent generation.
The synthesis of OPE scaffolds from TMS-acetylene 34, diiodobenzene 35 and acetate 31 has been recently adapted to the modular preparation of multichromophoric OPE-NDI hybrids.27 Deprotection of intermediate 36 prepared for the coupling of decaacid 37 with amines 26 and 27 to afford, after deprotection, the desired cationic and anionic OPE-RO20 and 21, respectively. The synthesis of POP- and OPE-NDI initiators and propagators 1–11 and 13–17 has been described previously.1,26,27,42
Cyclic voltammetry
Redox potentials were determined by cyclic voltammetry (CV) in dichloromethane under conditions used previously with the ferrocene/ferricenium couple (Fc/Fc+) as internal standard.25–27 The reversible NDI/NDI− reduction of monomeric RO regioiosmer (3,7)-25 occurred at a midpoint redox potential E0red1 = −1.25 V (Fig. 4).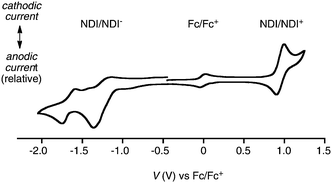 | ||
| Fig. 4 Cyclic voltammogram of (3,7)-25 in dichloromethane with the Fc/Fc+ couple as internal standard (scan rate, 100 mV s−1; supporting electrolyte, 100 mM Bu4NPF6; working electrode, Pt disk; counter electrode, Pt wire; reference electrode, Ag/AgCl). The CV of (2,6)-25 was roughly the same. | ||
The onset of the first reduction potential Ered1onset = −1.05 V was taken to calculate the energy level of the LUMO ELUMO = −3.75 eV against vacuum (Table 1, entry 1). This value was roughly identical for both regioisomers as well as the halogenated red RCl and RBr (Table 1, entries 1–3). The onset of the first oxidation potential Eox1onset = +0.95 V was used correspondingly to calculate the EHOMO = −5.75 eV (Table 1, entry 1). Consistent with the π-donating nature of the alkoxy substituent, this HOMO energy was 0.15 eV above the EHOMO = −5.90 eV of RCl and RBr (Table 1, entries 2 and 3).
| Entry | Cpdb | E ox1 onset/Vc | E 0 red1 onset/Vd | λ max1 onset/nme | HOMO/eVf | LUMO/eVg | E g CV/eVh | E g opt/eVi | Ref |
|---|---|---|---|---|---|---|---|---|---|
| a For original data and conditions, see Fig. 4, for other NDIs, see Fig. 1. b Given for (3,9)-25. c Onset of first oxidation potential in Volts against Fc/Fc+. d Onset of first reduction potential in Volts against Fc/Fc+. e Onset of longest wavelength absorption. f Energy of highest occupied molecular orbital against vacuum, from −4.8 eV for Fc/Fc+ minus Eox1onset. g Energy of the lowest unoccupied molecular orbital against vacuum, from −4.8 eV for Fc/Fc+ minus Ered1onset. h Electrochemical band gap EgCV (eV) = LUMO–HOMO. i Optical band gap Egopt (eV) = 1240/λmax1onset (nm). | |||||||||
| 1 | RO | 0.95 | −1.05 | 600 | −5.75 | −3.75 | 2.00 | 2.07 | |
| 2 | RCl | 1.11 | −1.02 | 570 | −5.91 | −3.78 | 2.13 | 2.18 | 25 |
| 3 | RBr | 1.10 | −1.05 | 570 | −5.90 | −3.75 | 2.15 | 2.18 | 27 |
| 4 | OPE | 0.80 | nd | 450 | −5.60 | −2.84c | nd | 2.76 | 27 |
| 5 | POP | 0.95 | nd | 365 | −5.75 | −2.35c | nd | 3.40 | 25 |
Within experimental uncertainties, the HOMO energy of RO chromophores was identical with that of POP scaffolds (Table 1, entry 5). This suggested that rod/stack PCS in POP-RO architectures would depend on the redox finetuning by the environment in supramolecular architectures (∼ ±0.5 eV), whereas the same process should be less critical in OPE-RO photosystems (ΔE = 0.15 eV, Table 1, entry 4). This comparison suggested that OPE-RO architectures should give better SHJ photosystems than POP-RO architectures.
Steady-state spectroscopy
The absorption spectra of RO25 in MeOH contained the band around 350 nm with a vibrational progression that is observed with all NDI chromophores due to a π–π* transition involving the NDI center (Fig. 5). The low-energy transition of the alkoxylated RO at 553 nm was clearly red-shifted compared to the halogenated RBr and RCl. The optical bandgap Egopt = +2.07 eV obtained from the onset of the charge-transfer band absorption at λmax1onset = 600 nm corresponded well with the electrochemical bandgap EgCV = +2.00 eV (Table 1, entry 1). The charge-transfer bands of regioisomers (2,6)-25 and (3,7)-25 in dichloromethane did not overlap and had maxima at 553 nm and 555 nm. The same small difference was reproduced in emission maxima at 581 nm and 583 nm.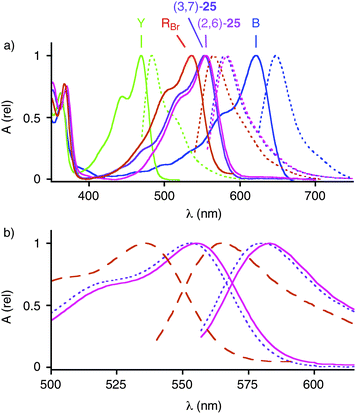 | ||
| Fig. 5 (a) Absorption (solid) and emission (dotted) spectra of the two regioisomers of RO, i.e., (2,6)-25 (magenta) and (3,7)-25 (violet), in dichloromethane compared to the corresponding Y (green), RBr and RCl (red, mixtures of regioisomers), and B (blue, data adapted from ref. 26). (b) Magnified absorption and emission spectra of (2,6)-25 (solid) and (3,7)-25 (dotted) compared to the corresponding RBr and RCl (dashed). | ||
Originating from locally-excited singlet state transition (S0 → NDI-LES), the low energy band at ∼550 nm of both POP-RO18 and OPE-RO20 was slightly broader and ∼170 cm−1 red-shifted compared to RO25 (Fig. 6). Attributed to excitonic interactions between the NDI units, similar effects have already been observed with other POP- and OPE-NDI systems.26,41,48 At high energy, POP-RO18 and OPE-RO20 showed additional absorptions at ∼320 nm and ∼400 nm originating from S0 → POP-LES and S0 → OPE-LES, respectively.
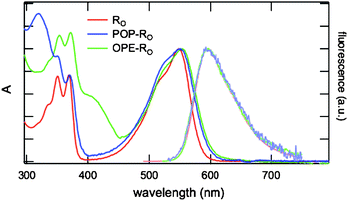 | ||
| Fig. 6 Normalized absorption and emission spectra of OPE-RO20 and POP-RO18 compared to RO25 in MeOH. | ||
The fluorescence spectra of all three compounds in MeOH obtained upon local NDI excitation were almost identical, with a maximum at 595 nm. The fluorescence quantum yield of monomeric RO25 (Φf = 0.57) was larger than that of yellow (Y, Φf = 0.08) and blue (B, Φf = 0.32) NDIs with two alkoxy or alkylamino substituents in the core (Table S1, Fig. 1). The poor Φf of Y has been ascribed to intermolecular hydrogen-bond assisted non-radiative deactivation.26 This process is most probably inhibited in B because of the formation of stronger intramolecular hydrogen bonds. Expected to be in between Y and B, the origin of the high Φf of RO is not understood.
Time-resolved fluorescence
The fluorescence decay of RO25 recorded by time-correlated single photon-counting (TCSPC) upon 469 nm excitation was monoexponential with a 10.5 ns lifetime (Table S1). This is longer than the lifetime found with B (8.4 ns), Y (2.6 ns) and RCl (2.3 ns) and agrees with the higher fluorescence quantum yield of RO. The fluorescence decay of POP-RO18 and OPE-RO20 was much faster than that of RO25, in agreement with the much smaller fluorescence quantum yields. TCSPC data of POP-RO18 exhibited biexponential behavior, that of OPE-RO20 a 10 ns decay only (Table S1). Fluorescence upconversion (FU) measurements upon 400 nm excitation of OPE-RO20 revealed additional decay components that are too fast to be resolved by TCSPC (Figure S8). Transient absorption (TA) measurements confirmed that the same is true for POP-RO18. These results pointed toward very efficient non-radiative deactivation pathways of the LES in POP-RO18 and OPE-RO20.Transient absorption measurements
The TA spectra of OPE-RO20 and POP-RO18 obtained after local NDI excitation at 550 nm were essentially identical (Fig. 7, top). At early time delays, they consisted of two positive bands, one around 500 nm with a shoulder at 530 nm and one at 670 nm, and one negative band at about 550 nm. During the first ∼10 ps, the 530 nm shoulder vanished, and a new positive band peaking at 610 nm appeared. Parallel increase of the 500 nm band and decrease of the 670 nm band was observed. From ∼20 ps onward, the TA spectral shape remained essentially constant but its intensity decayed to zero within ∼1.5 ns.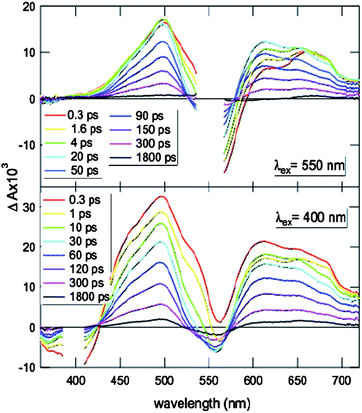 | ||
| Fig. 7 Transient absorption spectra recorded with the cationic OPE-RO20 in MeOH at different time delays after excitation at 550 nm (top) and at 400 nm (bottom) with a fs-laser pulse. | ||
This temporal variation of the TA spectra could be ascribed to the occurrence of a very fast charge separation (CS) process directly after excitation, followed by a slower charge recombination (CR) to the ground state. The 530 nm shoulder and the 670 nm band could be assigned to the absorption of the NDI-LES, as the TA spectrum of RO25 exhibited two positive bands at these wavelengths. The positive TA bands at 500 nm and above 600 nm were very similar to those measured with the radical anions of RCl.49 The TA spectra have been analyzed globally using multiexponential functions as described for OPE-B and OPE-RCl.41 This analysis revealed that for POP-RO18 and OPE-RO20, CS and CR are both biphasic (Table 2).
| Cpd | λ ex/nm | τ 1 | τ 2 | τ 3 | τ 4 | τ 5 | τ 6 |
|---|---|---|---|---|---|---|---|
| a The numbers in parantheses are tentative assignments according to the schemes depicted in Fig. 8. | |||||||
| 18 (POP-RO) | 550 (MeOH) | 1.1 ps (1, 2) | 9.4 ps (1, 2) | 81 ps (3, 4) | 430 ps (3, 4) | ||
| 20 (OPE-RO) | 550 (MeOH) | 1.9 ps (5) | 12.0 ps (5) | 93 ps (9) | ∼1 ns (9) | ||
| 38 (OPE-RO) | 550 (DMF) | 0.5 ps (5, 6) | 3.6 ps (5, 6) | 136 ps (9, 10) | 500 ps (9, 10) | ||
| 20 (OPE-RO) | 400 (MeOH) | 0.2 ps (7, 8) | 8.0 ps (5, 7, 8) | 60 ps (5) | 260 ps (10) | >2 ns (9) | |
| 38 (OPE-RO) | 400 (DMF) | 0.2 ps (7) | 0.4 ps (7, 8) | 2.4 ps (5, 8) | 19 ps (5) | 150 ps (9) | >1 ns (10) |
The TA spectra of POP-RO18 confirmed the formation of the RO radical anions in the charge-separated state (CSS), but the hole could not be localized. As POP and RO have the same oxidation potential, symmetry-breaking CS to the NDI˙+/NDI˙− CSS (pathway 1 in Fig. 8) or CS to the POP˙+/NDI˙− CSS (pathway 2) are both energetically possible. CR with POP-RO18 (81 and 430 ps) was faster than with the POP˙+/NDI˙− CSS of POP-RCl7 (100 ps and 1.1 ns) and slower than with the NDI˙+/NDI˙− CSS of POP-B 3 (45 ps).24,43
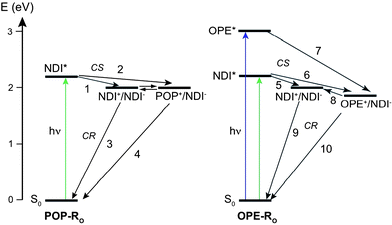 | ||
| Fig. 8 Energy level schemes pertaining to the relevant CS and CR processes in POP-RO18 (left) and OPE-RO20 (right). | ||
The TA spectra recorded with OPE-RO20 upon OPE excitation at 400 nm resulted in the population of the OPE-LES and, to a lesser extent, that of the NDI-LES (Fig. 7, bottom). The very early spectra exhibited a negative band at 400 nm that can be ascribed to the bleaching of the S0 → OPE-LES transition, and the two positive bands of the radical anion of RO. The dip around 550 nm, due to the bleaching of the S0 → NDI-LES transition, was larger than zero because of the OPE-LES population, which exhibited a broad absorption band in the visible.50 Over the few first picoseconds, the intensity of the positive band decayed partially, that in the 550 nm region became negative and the bleaching at 400 nm smaller. These changes could be assigned to the decay of the OPE-LES population by CS. After 1.8 ns, the bleaching at 400 nm had completely vanished, whereas the bands due to the RO anion could still be observed. Because of the presence of the RO anion bands and of the absence of OPE bleaching at 400 nm, this residual TA spectrum could be attributed to the NDI˙+/NDI˙− CSS.
Global analysis of the TA spectra required the sum of not less than five exponential functions (Table 2). From the decay associated spectra, it appeared that the 200 fs time constant can be assigned to CS from the OPE-LES to the OPE˙+/NDI˙− CSS (pathway 7 in Fig. 8). As part of the 400 nm bleaching recovered with the same time constant, one could conclude that a fraction of the OPE˙+/NDI˙− CSS population converts into the NDI˙+/NDI˙− CSS on a similar time scale (pathway 8). The 8 ps component could also be associated with CS and partial conversion to NDI˙+/NDI˙−. The 60 ps time constant was related to CS as well but not to the decay of the OPE bleach. Therefore, we assigned this component to CS from the NDI-LES directly populated upon 400 nm excitation to NDI˙+/NDI˙− (pathway 5). Finally, the last two time constants could be ascribed to CR. As the final stage of the OPE ground-state recovery occurred in 260 ps as well, this time constant could be interpreted as CR of the OPE˙+/NDI˙− CSS (pathway 10), whereas the longest time constant is due to the CR of the NDI˙+/NDI˙− CSS (pathway 9). In this long-lived CSS, the two charges are located on two remote NDIs as already observed with OPE-B,41 and CR first requires charge hopping to adjacent NDIs.
TA spectra measured with OPE-RO20 upon local NDI excitation at 550 nm did not exhibit a negative band at 420 nm (Fig. 7, top). This absence of the bleaching of the S0 → OPE-LES transition indicated that in this case, the OPE scaffold is not involved in the excited-state dynamics, i.e., the presence of the SHJ-incompatible NDI˙+/NDI˙− rather than the SHJ-compatible OPE˙+/NDI˙− CSS. This preference was surprising because the oxidation potential of OPE has been measured 150 mV below that of RO (Table 1, Fig. 8, right). This suggested that factors like the solvation energy, electrostatic interaction between the charged moieties, coupling between proximal NDIs, and so on might in this case invert this small difference in oxidation potential. However, the subtle difference between the involved HOMO levels suggested that other OPE-RO systems measured under different conditions could provide access to the OPE˙+/NDI˙− CSS. For this purpose, TA measurements of the cationic OPE-RO20 in MeOH (Fig. 7) were complemented by TA measurements of the neutral OPE-RO38 in DMF (Fig. 9).
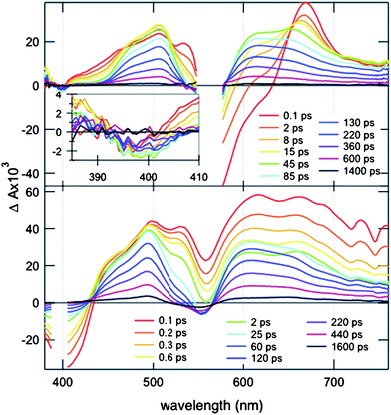 | ||
| Fig. 9 Transient absorption spectra recorded with the neutral OPE-RO38 in DMF at different time delays after excitation at 550 nm (top) and at 400 nm (bottom) with a fs-laser pulse. | ||
The TA spectra of the neutral OPE-RO38 obtained in DMF in response to OPE excitation at 400 nm (Fig. 9, bottom) were very similar to the ones of the cationic OPE-RO20 in MeOH (Fig. 7, bottom). Slightly shorter CSS lifetimes originated presumably from the non-protic nature of DMF rather than the negligible differences in polarity compared to MeOH (εs = 36.7 vs. 32.7, Table 2).
The TA spectra of the neutral OPE-RO38 obtained in DMF upon NDI excitation at 550 nm exhibited a negative band at 400 nm (Fig. 9, top) that was not present in the TA spectra of the cationic OPE-RO20 in MeOH (Fig. 7, top). This feature could be assigned to the bleaching of the S0 → OPE-LES transition and pointed to an involvement of the OPE unit in the CSS. The amplitude of this negative band was small, indicative of a relatively poor OPE˙+/NDI˙− CSS population. TA data could neither support nor exclude a possible conversion from OPE˙+/NDI˙− to NDI˙+/NDI˙− CSS (pathway 8).
The neutral OPE-RO38 was insoluble in MeOH. TA measurements with the cationic OPE-RO20 in DMF have been performed upon NDI excitation at 550 nm. Precise determination of CS and CR dynamics was not possible because poor solubility caused strong scattering of the pump light. However, the quality of the TA spectra was sufficient to show that the OPE bleach around 400 nm was missing. The OPE bleach observed with the neutral OPE-RO38 in DMF thus originated from the absence of charges in the NDIs sidechains rather than from the solvent. Conformational changes in response to reduced sidechain repulsion have been found previously to influence the optoelectronic properties of OPE-B systems significantly.41,42 Similar structural (rather than electric) effects were likely to account for the shift from NDI˙+/NDI˙− CSS to OPE˙+/NDI˙− CSS in response to reduced sidechain repulsion in OPE-RO systems. The TA experiments thus demonstrated that OPE-RO systems are compatible with OMARG-SHJ zipper architectures and provide access to photoinduced rod/stack charge separation with minimal losses in photonic energy.
Photocurrent generation
For zipper and LBL assembly, monolayers of POP-N 11, OPE-N 17 and lipoic acid 12 on freshly cleaned gold electrodes were prepared and characterized following previously reported procedures (Fig. 3).23–27 POP-RO architectures Au-11-(18-19-)n were zipped up by repetitive incubation of Au-11 initiators into dilute solutions of cationic and anionic POP-RO propagators 18 and 19. OPE-RO zippers Au-17-(20-21-)n were prepared analogously from Au-17 initiators with cationic and anionic OPE-RO propagators 20 and 21. To prepare the presumably less ordered LBL architectures Au-12-(18-19-)n and Au-12-(20-21-)n, anionic Au-12 monolayers without the initiators of zipper assembly were used.Photocurrent generation was evaluated with the photosystems as electron acceptors, a Pt electrode as cathode, and triethanolamine (TEOA) as electron donor. Photocurrent generation and termination by all explored systems was instantaneous, and the obtained currents were stable.23,25 The photocurrents generated by the POP-RO zipper Au-11-(18-19-)n increased about linearly with increasing number of layers (Fig. 10, ○). Saturation was reached at a critical thickness1 of 10 layers and a quite modest Jmax = 3.8 μA cm−2 (Table 3, entry 3). The corresponding POP-RO LBL architectures Au-12-(18-19-)n generated clearly less photocurrent (Fig. 10, □). With POP-RO systems, zipper assembly was clearly slower than LBL assembly. J–L profiles of the latter were roughly independent of incubation times. In clear contrast, J–L profiles of zipper assembly obtained with incubation for one day per layer (Fig. 10, ●) were not much better than LBL profiles and clearly weaker than the ones obtained at completion after three days per layer (Fig. 10, ○). These differences in kinetics26 and photocurrent generation added to corroborative experimental evidence available that zipper assembly exists and matters for function.
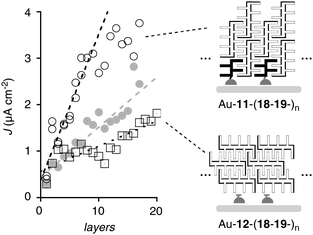 | ||
| Fig. 10 J–L Profiles of POP-RO zipper Au-11-(18-19-)n with incubation time of 3 days (○) and 1 day (●) per layer compared to LBL assembly Au-12-(18-19-)n (□), lines are added to guide the eye. | ||
| Entry | Architectureb | Designationb | L c (layers)c |
J
max/μA cm−2d |
J
SC/μAcm−2e,f |
V OC/Ve,g | FF , |
|---|---|---|---|---|---|---|---|
| a J–V data are given for n = 9 for OPE and n = 7 for POP, input power Pin = 66 mW cm−2 except for POP LBL with Pin = 100 mW cm−2. b See Fig. 1–3 and text for details. c Critical thickness in J–L curves, from Fig. 10 and 11. d Maximal photocurrent density in J–L curves, from Fig. 10 and 11. e J–V data, from Fig. 12. f Short circuit current density. g Open circuit voltage. h Fill factor FF = maximum power/(VOC × JSC) = (Vm × Jm)/(VOC × JSC), Vm = voltage at maximal power Pmax, Jm = photocurrent density at Pmax. | |||||||
| 1 | Au-17-(20-21-)n-20 | OPE-RO zipper | 20 | 16.0 | 15.3 | −0.40 | 0.55 |
| 2 | Au-12-(20-21-)n-20 | OPE-RO LBL | 20 | 6.0 | 5.2 | −0.39 | 0.54 |
| 3 | Au-11-(18-19-)n-18 | POP-RO zipper | 10 | 3.8 | 2.6 | −0.38 | 0.56 |
| 4 | Au-12-(18-19-)n | POP-RO LBL | — | 1.6 | 1.1 | −0.29 | 0.53 |
The J–L profile of OPE-RO zipper Au-17-(20-21-)n was characterized by a critical thickness of around 20 layers and a maximal photocurrent density Jmax = 16.0 μA cm−2 (Fig. 11, ○; Table 3, entry 1). The corresponding OPE-RO LBL assembly Au-12-(20-21-)n had the same critical thickness and produced, with Jmax = 6.0 μA cm−2, much less photocurrent (Fig. 11, □; Table 3, entry 2). Both photocurrent generation and critical thickness of OPE-RO zipper Au-17-(20-21-)n were clearly better than that with POP-RO zipper Au-11-(18-19-)n (Table 3, entries 1 vs. 3). Superior performance of OPE compared to POP zippers has been observed previously for RBr and B systems.27,42 It originates from several contributions including photocurrent generation by OPEs as well as topological matching.27
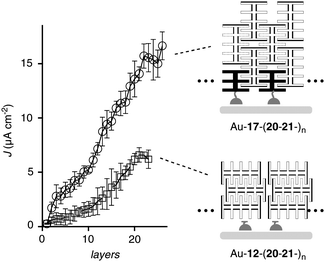 | ||
| Fig. 11 J–L Profiles of OPE-RO zipper Au-17-(20-21-)n (○) and LBL assembly Au-12-(20-21-)n (□). | ||
The J–V profile of OPE-RO zipper Au-17-(20-21-)9-20 revealed a strongly non-linear dependence of photocurrent generation on voltage (Fig. 12, ●; Table 3, entry 1). This important behavior was reflected in a fill factor FF = 0.55. Despite much weaker short-circuit photocurrent density JSC, POP-RO zipper Au-11-(18-19-)9-18 had the same fill factor (Fig. 12, ○; Table 3, entry 3). Almost the same was true for the corresponding, otherwise clearly less active, LBL architectures (Table 3, entries 2 and 4). These comparably high fill factors were in the range of those observed previously with other OPE-NDI zippers (OPE-RBr, FF = 0.61;27 OPE-B, FF = 0.5242) and some but not all POP-NDI zippers (POP-RBr, FF = 0.61;27 POP-Y, FF = 0.53;26 POP-B, FF = 0.3442). Particularly interesting are the high fill factors at low photocurrents observed with POP-RO zippers, because decreasing FF with decreasing JSC is more frequently observed, independent of the architecture of the photosystem (e.g., OPE-B, FF = 0.52 vs. POP–B, FF = 0.34).42 High fill factors have been considered, together with critical thickness, as indication of high organization of artificial photosystems.1,9 Relatively high FF at relatively low JSC with RO photosystems other than POP-RO suggested that poor activity does not originate from disorganization but from the malfunctioning of well-organized architectures.
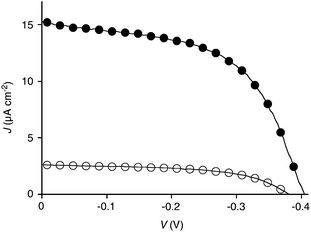 | ||
| Fig. 12 J–V profile of POP-RO zipper Au-11-(18-19-)7-18 (○) and OPE-RO zipper Au-17-(20-21-)9-20 (●), see Table 1 for data analysis. | ||
The action spectrum of the red POP zipper Au-11-(18-19-)n revealed that the pinkish NDI chromophore RO is capable of generating a photocurrent (Fig. 13b, ●). The action spectrum of POP-RO zippers was overlayed with previously reported action spectra and normalized at the common POP π–π* transition at 350 nm. Photocurrent generation by photoexcited NDI chromophores decreased with Y > RBr > RO ≫ B (Fig. 13b). Upon normalization at 400 nm of the OPE π–π* transition, the same RBr > RO ≫ B trend was found with OPE-NDI zippers (Fig. 13c).
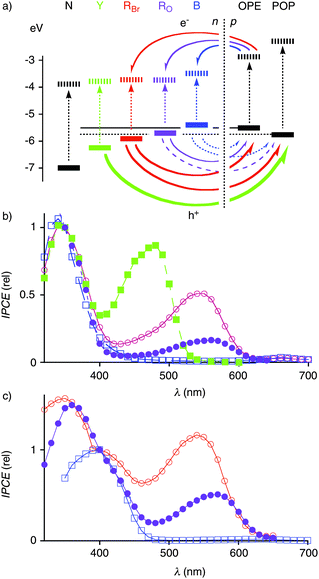 | ||
| Fig. 13 Action spectrum of POP-RO zippers (b, ●, violet, Au-11-(18-19-)7-18) and OPE-RO zippers (c, ●, violet, Au-17-(20-21-)7-20) compared to the corresponding POP (b) and OPE (c) zippers with RBr (○, red),27 B (□, blue),42 and Y (■, green)26 NDIs. Incident photon-to-current conversion efficiencies (IPCEs) are normalized at (b) 350 nm or (c) 400 nm. (a) Involved energy levels (see Fig. 1) with favorable (bold), possible (solid), less favorable (dashed) and unfavorable (dotted) photoinduced rod/stack electron (e−) and hole (h+) transfers (arrows). | ||
Taken together, the ability of POP- and OPE-NDI zippers to generate photocurrent increased with decreasing HOMO energy of the NDI chromophore (Fig. 13a). This remarkably consistent series suggested that photoinduced hole transfer from the NDI to the OPE or POP rod is of high importance for photocurrent generation. Blue NDIs with HOMO levels above OPEs and POPs essentially failed to generate photocurrent.42 Yellow NDIs with low-lying HOMOs generated most photocurrent.26 Among the red NDIs RO and RBr, the higher HOMO of the former was correctly reflected in a reduced ability to generate photocurrent.
Given the complexity of the system, it is understood that the clean dependence on the HOMO levels of hole donors and acceptors does not exclude other explanations of the trends found for photocurrent generation. Important contributions related to the TEOA carriers, for instance, remain possible. With regard to such concerns, it is important to note that the RO chromophores in OPE-RO zippers generated more photocurrent than the ones in POP-RO zippers (Fig. 13b vs. 13c, ●). This difference was consistent with the functional importance of favorable hole transfer from the excited RO to the better OPE acceptor. Photocurrent generation by RO in POP-RO zippers was in clear contrast to the nearly inactive B in POP-B zippers and implied that photoinduced hole transfer from excited RO to POP acceptors remains possible. Validity of these interpretations was corroborated by TA spectroscopy, where the formation of OPE˙+/RO˙− CSS could be observed besides the otherwise preferred RO˙+/RO˙− CSS when reducing the charge repulsion between the NDI sidechains. This conditional detectability of OPE˙+/RO˙− CSS suggested that in OPE-RO, the inevitable energy loss by rod/stack PCS is as small as possible.
Conclusions
The objective of this study was to explore the usefulness of red, electron-transporting NDI chromophores with one alkoxy and one alkylamino substituent in the core (i.e. RO) to serve in functional rod/stack surface architectures. We report that POP-RO and OPE-RO systems form functional zipper assemblies. Increased critical thickness with OPE-RO compared to POP-RO systems confirm the importance of topological matching for zipper assembly. With POP-RO zippers producing clearly more photocurrent than their LBL assemblies, we conclude that non-halogenated RO chromophores are less sensitive to topological mismatch than their halogenated red counterparts RBr.27 RO chromophores are thus more robust and adaptable for applications toward complex zipper architectures, including future OMARG-SHJs.1 High organization in POP-RO and OPE-RO architectures is supported by invariably high fill factors between 0.53 and 0.56.9Transient absorption measurements demonstrate that hole injection into OPE acceptors after RO excitation can occur. Decreasing photocurrent generation with increasing HOMO levels in Y > RBr > RO > B in OPE and POP series supports the functional importance of this SHJ-type hole rod/stack PCS, although other explanations of this trend cannot be fully excluded at this stage. Gradually decreasing photocurrent generation with increasingly unfavorable photoinduced rod/stack charge separation in OPE-RO systems illustrates that hole transfer decreases gradually (rather than abruptly) with increasingly subtle HOMO differences. We conclude that the optoelectronic finetuning of the HOMO levels of RO donors and OPE acceptors should be nearly perfect to assure SHJ-type rod/stack PCS with minimal losses in photonic energy. The alternative route to SHJs by electron injection from excited OPEs into surrounding RO acceptors is as unproblematic as with RBr and B acceptors.
Taken together, these results demonstrate that the mismatch-sensitive POP-RBr and OPE-RBr modules can be replaced by POP-RO and OPE-RO to give more robust zipper architectures where SHJ-compatible rod/stack PCS can occur with minimal losses in photonic energy and without complications from heavy atom effects. These advantages come at the cost of reduced photocurrent generation by the NDI chromophores. This means that eventual gains in open circuit voltage from maximized optoelectronic finetuning might risk coinciding with losses in short-circuit current. Nevertheless, we conclude that the RO chromophores are a good alternative to RBr chromophores to be integrated as valid components into future OMARG-SHJ photosystems made by zipper assembly. Synthetic efforts toward this biomimetic ideal are ongoing. Current efforts focus on high-energy acceptors in the hole transporting channel that can integrate blue NDI donors on the one hand and OPE donors on the other. They will be needed to build complete OMARG-SHJ photosystems with three-component gradients in both hole and electron-transporting pathways.
Acknowledgements
We thank A. Perez-Velasco, S. Sakurai, D.-H. Tran and L. Maffiolo for contributions to synthesis, D. Jeannerat, A. Pinto and S. Grass for NMR measurements, P. Perrottet N. Oudry and G. Hopfgartner for MS measurements, and the University of Geneva and the Swiss NSF for financial support.References
- R. Bhosale, J. Misek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- C. Ma, M. Fonrodona, M. C. Schikora, M. M. Wienk, R. A. J. Janssen and P. Bäuerle, Adv. Funct. Mater., 2008, 18, 3323–3331 CrossRef CAS.
- J.-F. Eckert, J.-F. Nicoud, J.-F. Nierengarten, S.-G. Liu, L. Echegoyen, N. Armaroli, F. Barigelletti, L. Ouali, V. Krasnikov and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 7467–7479 CrossRef CAS.
- V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003 Search PubMed.
- F. Yang, M. Shtein and S. Forrest, Nat. Mater., 2005, 4, 37–41 CrossRef CAS.
- F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C.-C. You, P. Jonkheijm, J. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611–10618 CrossRef.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS.
- H. J. Snaith, G. L. Whiting, B. Sun, N. C. Greenham, W. T. S. Huck and R. H. Friend, Nano Lett., 2005, 5, 1653–1657 CrossRef CAS.
- R. C. Shallcross, G. D. D'Ambruoso, B. D. Korth, H. K. Hall, Z. Zheng, J. Pyun and N. R. Armstrong, J. Am. Chem. Soc., 2007, 129, 11310–11311 CrossRef CAS.
- E. Hwang, K. M. N. de Silva, C. B. Seevers, J.-R. Li, J. C. Garno and E. E. Nesterov, Langmuir, 2008, 24, 9700–9706 CrossRef CAS.
- C.-H. Huang, N. D. McClenaghan, A. Kuhn, G. Bravic and D. M. Bassani, Tetrahedron, 2006, 62, 2050–2059 CrossRef CAS.
- M. Morisue, S. Yamatsu, N. Haruta and Y. Kobuke, Chem.–Eur. J., 2005, 11, 5563–5574 CrossRef CAS.
- A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198–3200 CrossRef CAS.
- A. B. F. Martinson, A. M. Massari, S. J. Lee, R. W. Gurney, K. E. Splan, J. T. Hupp and S. T. Nguyen, J. Electrochem. Soc., 2006, 153, A527–A532 CrossRef CAS.
- F. B. Abdelrazzaq, R. C. Kwong and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 4796–4803 CrossRef CAS.
- D. M. Guldi, J. Phys. Chem. B, 2005, 109, 11432–11441 CrossRef CAS.
- N. Martín, L. Sánchez, M. Ángeles Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef CAS.
- S. Sista, Y. Yao, Y. Yang, M. L. Tang and Z. Bao, Appl. Phys. Lett., 2007, 91, 223508 CrossRef.
- N. Sakai, A. L. Sisson, T. Bürgi and S. Matile, J. Am. Chem. Soc., 2007, 129, 15758–15759 CrossRef CAS.
- A. L. Sisson, N. Sakai, N. Banerji, A. Fürstenberg, E. Vauthey and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 3727–3729 CrossRef CAS.
- N. Sakai, R. S. K. Kishore and S. Matile, Org. Biomol. Chem., 2008, 6, 3970–3976 RSC.
- R. S. K. Kishore, O. Kel, N. Banerji, D. Emery, G. Bollot, J. Mareda, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2009, 131, 11106–11116 CrossRef CAS.
- R. Bhosale, A. Perez-Velasco, V. Ravikumar, R. S. K. Kishore, O. Kel, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, T. Sawada, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2009, 48, 6461–6464 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070–8075 CrossRef.
- S. Gabutti, S. Schaffner, M. Neuburger, M. Fischer, G. Schäfer and M. Mayor, Org. Biomol. Chem., 2009, 7, 3222–3229 RSC.
- S. Chopin, F. Chaignon, E. Blart and F. Odobel, J. Mater. Chem., 2007, 17, 4139–4146 RSC.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS.
- J. Mareda and S. Matile, Chem.–Eur. J., 2009, 15, 28–37 CrossRef CAS.
- W. Hu, N. Zhu, W. Tang and D. Zhao, Org. Lett., 2008, 10, 2669–2672 CrossRef.
- J. Kim and T. M. Swager, Nature, 2001, 411, 1030–1034 CrossRef CAS.
- S. A. McFarland and N. S. Finney, J. Am. Chem. Soc., 2002, 124, 1178–1179 CrossRef CAS.
- S. B. Sachs, S. P. Dudek, R. P. Hsung, L. R. Sita, J. F. Smalley, M. D. Newton, S. W. Feldberg and C. E. D. Chidsey, J. Am. Chem. Soc., 1997, 119, 10563–10564 CrossRef CAS.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847–5864 RSC.
- R. Huber, T. M. Gonzlez, S. Wu, M. Langer, S. Grunder, V. Horhoiu, M. Mayor, M. R. Bryce, C. Wang, R. Jitchati, C. Schoenenberger and M. Calame, J. Am. Chem. Soc., 2008, 130, 1080–1084 CrossRef CAS.
- N. Banerji, G. Duvanel, A. Perez-Velasco, S. Maity, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. A, 2009, 113, 8202–8212 CrossRef CAS.
- S. Maity, R. Bhosale, N. Banerji, E. Vauthey, N. Sakai and S. Matile, Org. Biomol. Chem., 2010, 8, 1052–1057 RSC.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS.
- P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS.
- F. Würthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem.–Eur. J., 2002, 8, 4742–4750 CrossRef CAS.
- R. S. K. Kishore, V. Ravikumar, G. Bernardinelli, N. Sakai and S. Matile, J. Org. Chem., 2008, 73, 738–740 CrossRef CAS.
- N. Sakai, N. Majumdar and S. Matile, J. Am. Chem. Soc., 1999, 121, 4294–4295 CrossRef CAS.
- N. Banerji, A. Fürstenberg, S. Bhosale, A. L. Sisson, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. B, 2008, 112, 8912–8922 CrossRef CAS.
- G. Duvanel, J. Grilj, A. Schuwey, A. Gossauer and E. Vauthey, Photochem. Photobiol. Sci., 2007, 6, 956–963 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c0sc00177e |
| This journal is © The Royal Society of Chemistry 2010 |