
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers†
F. F.
Wagner
a,
Y.-L.
Zhang
a,
D. M.
Fass
ac,
N.
Joseph
ab,
J. P.
Gale
a,
M.
Weïwer
a,
P.
McCarren
a,
S. L.
Fisher
c,
T.
Kaya
a,
W.-N.
Zhao
ad,
S. A.
Reis
ad,
K. M.
Hennig
ad,
M.
Thomas
a,
B. C.
Lemercier
a,
M. C.
Lewis
a,
J. S.
Guan
ab,
M. P.
Moyer
a,
E.
Scolnick
a,
S. J.
Haggarty
ad,
L.-H.
Tsai
ab and
E. B.
Holson
*a
aStanley Center for Psychiatric Research, Broad Institute of Harvard and MIT, 415 Main Street, Cambridge, Massachusetts, USA. E-mail: edholson@broadinstitute.org
bPicower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA
cSL Fisher Consulting, LLC, PO Box 3052, Framingham, Massachusetts, USA
dChemical Neurobiology Laboratory, Center for Human Genetic Research, Massachusetts General Hospital, Department of Neurology and Psychiatry, Harvard Medical School, Boston, Massachusetts, USA
First published on 9th October 2014
Abstract
Aiming towards the development of novel nootropic therapeutics to address the cognitive impairment common to a range of brain disorders, we set out to develop highly selective small molecule inhibitors of HDAC2, a chromatin modifying histone deacetylase implicated in memory formation and synaptic plasticity. Novel ortho-aminoanilide inhibitors were designed and evaluated for their ability to selectively inhibit HDAC2 versus the other Class I HDACs. Kinetic and thermodynamic binding properties were essential elements of our design strategy and two novel classes of ortho-aminoanilides, that exhibit kinetic selectivity (biased residence time) for HDAC2 versus the highly homologous isoform HDAC1, were identified. These kinetically selective HDAC2 inhibitors (BRD6688 and BRD4884) increased H4K12 and H3K9 histone acetylation in primary mouse neuronal cell culture assays, in the hippocampus of CK-p25 mice, a model of neurodegenerative disease, and rescued the associated memory deficits of these mice in a cognition behavioural model. These studies demonstrate for the first time that selective pharmacological inhibition of HDAC2 is feasible and that inhibition of the catalytic activity of this enzyme may serve as a therapeutic approach towards enhancing the learning and memory processes that are affected in many neurological and psychiatric disorders.
Introduction
Mounting evidence, generated over the past decade, supports the critical role of chromatin modification and gene expression regulation in the molecular mechanisms underlying synaptic plasticity and memory formation.1 Dysregulation of these neurobiological processes manifest as a variety of cognitive phenotypes in a host of diseases including Alzheimer's disease (neurodegenerative2), schizophrenia3 (psychiatric), post-traumatic stress disorder (PTSD)4 (psychiatric), Rubinstein–Taybi and Rett's syndrome5 (intellectual disability). The learning and/or memory impairments associated with these disorders represent a profound unmet medical need that is not effectively ameliorated by current approved treatments. According to the 2014 Alzheimer's disease facts and figures report, the prevalence of AD alone is estimated to triple by 2050 and affect more than 13 million individuals in the United States. New treatments which focus beyond the slowing of disease progression in AD and are more broadly applicable across disease states are sorely needed. Because learning and memory processes require active gene transcription and subsequent protein synthesis to establish long-lasting changes in synapses, biological targets which affect gene expression are attractive for pharmacological intervention. Several chromatin modifying enzymes have been implicated in the neurobiology of learning and memory, in particular, histone deacetylases (HDACs).1,6 HDACs are responsible for catalyzing the posttranslational hydrolysis of acetyl groups from the ε-nitrogen of lysine residues located on histone as well as non-histone proteins.7–9 The metal-dependent isoforms are categorized as follows: Class I (HDACs 1, 2, 3 and 8), Class IIa (HDACs 4, 5, 7, 9), Class IIb (HDACs 6, 10) and Class IV (HDAC11).7 The dysregulation of histone acetylation is a feature associated with a range of neurological disorders.6 For example, Rubinstein–Taybi Syndrome (RTS), a rare human genetic disorder, is caused by mutations in the histone acetyltransferase (HAT) domain of the CREB-binding protein (CBP) gene.10 This loss of function mutation leads to a hypoacetylation state, in transgenic mice, that phenocopies cognitive deficits observed in humans. As a therapeutic proof of principle, the hypoacetylation in brain and the corresponding cognitive deficits in these mice can be rescued through the administration of SAHA, a non-selective Class I, IIb HDAC inhibitor. Subsequently, several groups demonstrated that administration of non-selective inhibitors, primarily SAHA and the Class I inhibitor sodium butyrate, can rescue the cognitive deficits in learning and memory behavioral paradigms for a variety of transgenic mouse models.11–15 Most recently, Gräff et al. show that treatment with CI-994, an HDAC1, 2 and 3 inhibitor, triggers the upregulation of a key set of neuroplasticity-related genes and was efficacious in fear extinction models of PTSD.4 A key question underlying the effects of these non-selective HDAC inhibitors is whether they are driven by the inhibition of a single or a combination of HDAC isoforms.Among the Class I and Class IIb isoforms, knockout and/or over-expression transgenic mouse models of HDAC2,12,16 HDAC315 and HDAC611,13 have demonstrated that loss of function of these individual isoforms can enhance memory and synaptic plasticity. While selective inhibitors of HDAC3 and HDAC6 have been described and in some cases demonstrated in vivo efficacy in mouse models of learning and memory, there are no such tools available for probing the selective inhibition of HDAC2 in the brain.17 Additionally, Tsai and co-workers demonstrated that HDAC1 activity may be neuroprotective,18 reinforcing the importance of selective inhibition within the class I isoforms. Intrigued by the opportunity for pharmacological intervention in psychiatric diseases characterized by a cognitive impairment component, and the increasing evidence implicating the role of HDAC2 in learning and memory, we set out to identify selective small molecule inhibitors of HDAC2.
Results and discussion
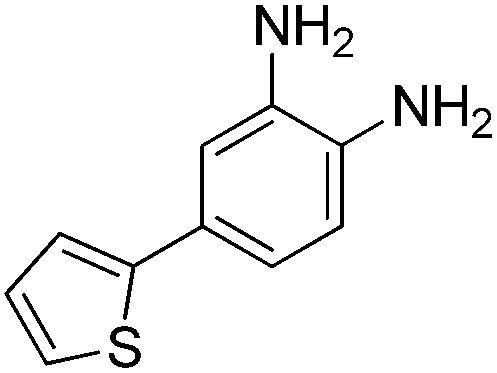
The development of highly potent and isoform selective HDAC inhibitors is critical not only to refine our understanding regarding the relevant isoform(s) for on-target efficacy but also to mitigate potential mechanism-based, dose-limiting side effects (thrombocytopenia, fatigue) caused by the inhibition of multiple HDACs, particularly HDACs 1 and 2.19 Among the Class I HDACs (HDACs 1, 2, 3 and 8), HDAC 1 and 2 share the highest overall sequence similarity (86%) and display 95% similarity within the Zn2+ catalytic binding domain.7 At the outset, we believed imparting sufficient selectivity between these two highly similar isoforms presented the greatest chemical challenge for small molecule binders targeting the HDAC catalytic binding domain. As part of our design strategy, we emphasized the kinetic (residence time) and thermodynamic binding properties of our inhibitors for HDACs 1, 2 and 3. Binding kinetics and residence times are important considerations when developing therapeutics.20 Compound residence time at the target of interest can dictate efficacy while its residence time at homologous target(s) could affect potential adverse effects. Ideally, a selective HDAC2 inhibitor would demonstrate both thermodynamic (Ki or IC50 values) and kinetic selectivity (residence time) favoring HDAC2. Another major challenge in CNS drug discovery, highlighted by the pharmacokinetic shortcomings of SAHA, is the efficient delivery of small molecules across the blood brain barrier (BBB). Consequently, our inhibitor design hinged on a multi parametric optimization of highly brain penetrant and selective inhibitors of HDAC2 versus all other Zn-dependent HDACs, paying particular attention to HDAC1.While there are several chemical classes of HDACi, we chose to focus our medicinal chemistry efforts on the ortho-aminoanilide class of inhibitors. Ortho-aminoanilides, exemplified by CI-994 (Table 1), are sub-Class I selective, inhibiting only HDAC1, 2 and 3, with no activity towards HDAC8 or the Class IIa and IIb HDAC isoforms (ESI Table 1†). Several groups have described HDAC1, 2 selective ortho-aminoanilides, exemplified by compound 1 (Table 1).21–23 The C-5 thiophene moiety, in compound 1, occupies a 14 Å internal cavity in HDACs 1 and 2 leading to improved selectivity and potency for these two isoforms. In addition, ortho-aminoanilides display slow binding kinetics.24–28 For example, compound 1 displays potent in vitro inhibition towards human recombinant HDACs 1 and 2 with pseudo-irreversible binding kinetics (Table 1; residence time determined through progression curve analysis for HDAC catalyzed deacetylation at various concentrations of inhibitor, see ESI†). Finally, ortho-aminoanilides are highly synthetically tractable and possess more desirable pharmacokinetic properties than other known HDAC inhibitor chemotypes.17,29
|
|
HDAC isoform inhibition IC50a (μM) | Ligand efficiencyb (ligE) | |||
|---|---|---|---|---|---|
| Compound | Structure | HDAC1 | HDAC2 | HDAC3 | HDAC1/HDAC2 |
a Values are the mean of a minimum of two experiments. Data are shown as IC50 values in μM ± standard deviation. Compounds were tested in duplicate in a 12-point dose curve with 3-fold serial dilution starting from 33.33 μM.
b Ligand efficiency (ligE) = (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50)/number of non-hydrogen atoms. IC50)/number of non-hydrogen atoms.
|
|||||
| Cl-994 |
|
0.041 ± 0.012 | 0.147 ± 0.066 | 0.046 ± 0.018 | 0.37/0.34 |
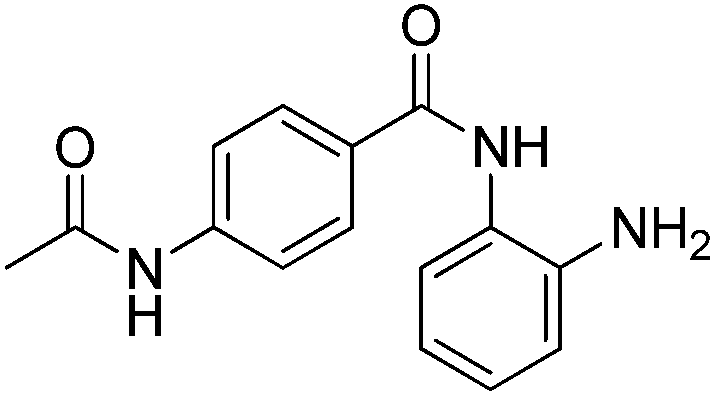
| 1 |
|
0.001 ± 0.001 | 0.013 ± 0.009 | 0.398 ± 0.105 | 0.36/0.32 |
| Residence time T 1/2 (min) | |||||
| >2400 | ∼4800 | ∼1200 | |||
| 2 |
|
0.023 ± 0.008 | 0.129 ± 0.006 | 1.68 ± 0.26 | 0.36/0.33 |
| 3 |
|
0.355 ± 0.012 | 8.71 ± 3.16 | 0.665 ± 0.063 | 0.40/0.32 |
| 4 |
|
>33.33 | >33.33 | >33.33 | |
In an effort to understand the contribution of the core binding motifs in 1 and maximize ligand efficiency in our design, we set out to identify the minimal pharmacophoric elements that confer potency for HDAC2.30 Starting from 1 (ligand efficiency (ligE) = 0.32 for HDAC2) we designed a series of truncated analogs starting from the solvent exposed acetamide motif (Table 1). Removal of the capping acetamide group provided compound 2, which retained moderate to good potency for HDACs 1, 2 and 3 (2, IC50 = 0.023 μM, 0.129 μM and 1.68 μM, respectively). Truncating further by removing the phenyl linking motif provided compound 3, which displayed weak to moderate potency for HDACs 1, 2 and 3 (3, IC50 = 0.335 μM; 8.71 μM and 0.665 μM, respectively). Accordingly, the ligand efficiencies of this truncated series for HDAC2 remains high (≥0.32). Finally, the biaryl-dianiline 4 displayed no inhibitory activity towards HDACs 1, 2 and 3. Intrigued by the ability of the highly efficient small molecule ligand 3 to bind HDAC1, 2 and 3, we performed molecular docking simulations into HDAC2 (3MAX structure)21 (Fig. 1A). Compound 3 achieves optimal chelation geometry establishing an intricate network of H-bonds with His145, His146 and Gly 154 (Fig. 1A and B). The docked structure demonstrates that the methyl amide is not only accommodated but provides a rigid vector aligned with the hydrophobic 11 Å channel (Fig. 1A) leading to the solvent exposed surface (Fig. 1B). On the basis of this model, we speculated that sp3-rich substituents projecting along this molecular trajectory would provide novel chemotypes and impart structure–activity relationships which were largely unexplored.
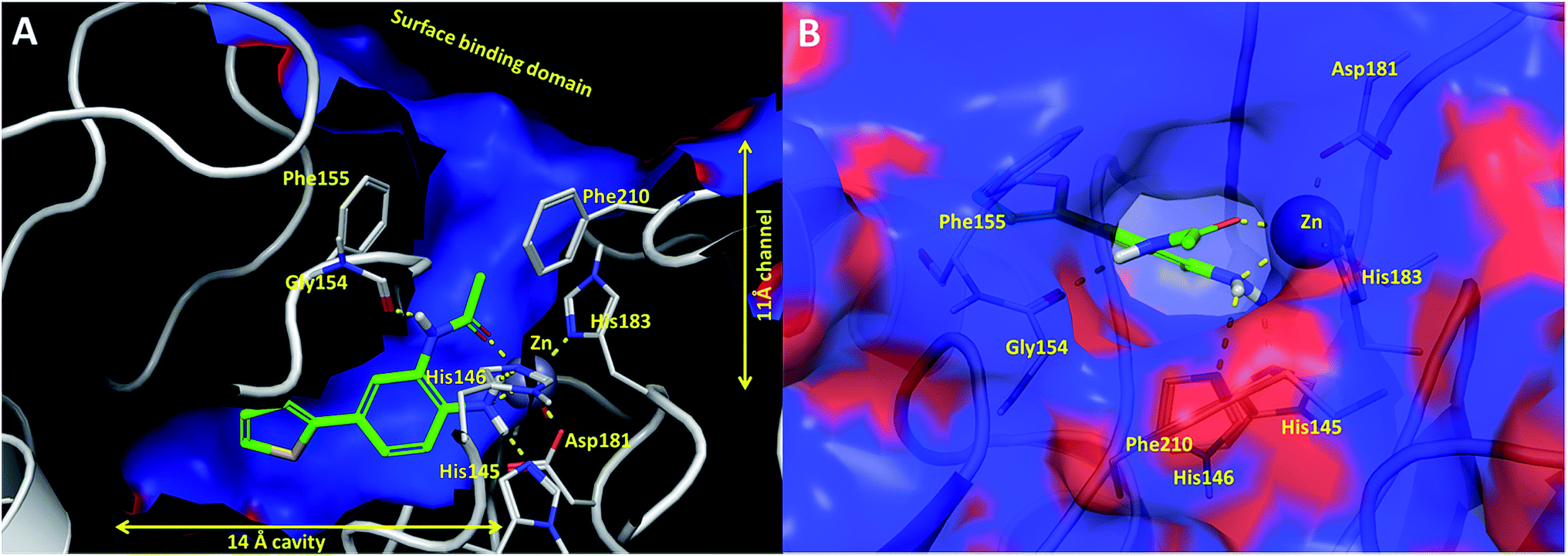 | ||
| Fig. 1 (A) Cross sectional view of compound 3 docked into catalytic binding domain of HDAC2 (B) surface view of compound 3 docked into HDAC2 showing the trajectory of the sp3 methyl amide into the 11 Å channel. Hydrogen bonding interactions are shown as yellow dotted lines. The electrostatic surface of HDAC2: blue = hydrophobic regions; red = negatively charged regions. | ||
While other sp3-linked ortho-aminoanilides have been described, they possess extended linker and capping groups that project beyond the 11 Å channel.31 We chose to focus our Structure Activity Relationship (SAR) efforts on the linker portion of the molecule occupying the 11 Å channel. To modulate compound properties (both physicochemical and binding kinetics), we chose three 14 Å internal cavity motifs: a 2-thienyl and p-fluorophenyl group, both of which are hydrophobic, and a more hydrophilic 4-pyridyl group.
Encouraged by the initial results with acetyl compound 3, we were inspired to chemically map the 3D topography of the linker region visibly available in our computational model (Fig. 1) to probe linker effects on potency, selectivity and kinetic binding, as well as the interplay with 14 Å internal cavity motifs. Using small sp3 rich linker groups (non-aromatic), we systematically explored the 11 Å channel (Table 2).
|
|
HDAC isoform inhibition IC50a (μM) | [Brain]/[plasma] and brain free fractionb (%) | ||||
|---|---|---|---|---|---|---|
| Compound | R1 group | R2 group | HDAC1 | HDAC2 | HDAC3 | |
| a Values are the mean of a minimum of two experiments. Data are shown as IC50 values in μM ± standard deviation. Compounds were tested in duplicate in a 12-point dose curve with 3-fold serial dilution starting from 33.33 μM. nd = not determined. b Brain free fraction estimated based on brain tissue binding experiments. | ||||||
| 5 |
|
|
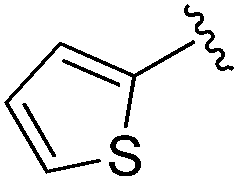
0.059 ± 0.015 | 0.261 ± 0.140 | 0.949 ± 0.034 | nd |
| 6 |
|
|
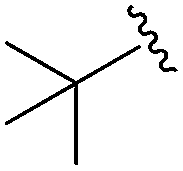
4.24 ± 0.257 | 3.13 ± 0.492 | 25.0 ± 1.71 | nd |
| 7 |
|
|
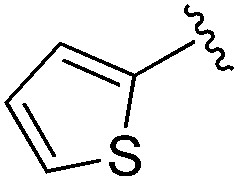
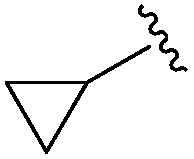
0.072 ± 0.029 | 0.086 ± 0.057 | 0.350 ± 0.018 | nd |
| 8 |
|

|
0.020 ± 0.003 | 0.131 ± 0.015 | 0.548 ± 0.162 | nd |
| 9 |
|
|
0.011 ± 0.005 | 0.095 ± 0.061 | 0.635 ± 0.308 | nd |
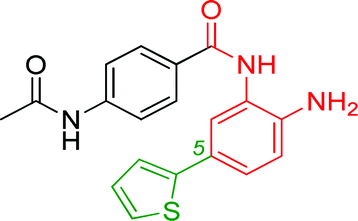
| BRD2283 |
|
|
0.003 ± 0.002 | 0.054 ± 0.016 | 0.604 ± 0.039 | nd |
| 10 |
|
|
0.021 ± 0.005 | 0.079 ± 0.042 | 1.01 ± 0.16 | nd |
| 11 |
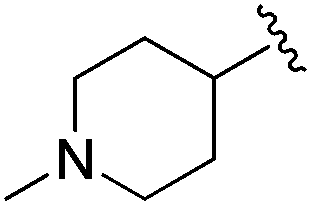
|
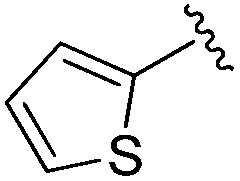
|
11.9 ± 1.17 | 13.23 ± 0.45 | >33.33 | nd |
| BRD3349 |
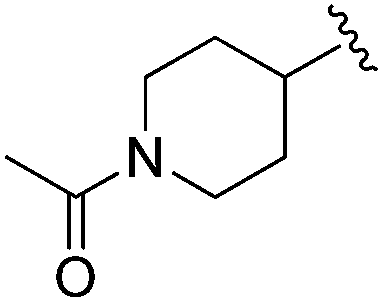
|
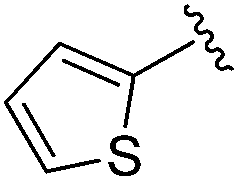
|
0.011 ± 0.003 | 0.049 ± 0.005 | 2.78 ± 0.02 | 0.04 |
| BRD4884 |
|
|
0.029 ± 0.012 | 0.062 ± 0.031 | 1.09 ± 0.38 | 1.29 |
| Residence time T 1/2 (min) | ||||||
| 20 | 143 | 257 | 6% | |||
| 12 |
|
|
2.36 ± 0.151 | 1.10 ± 0.019 | >33.33 | nd |
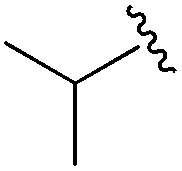
An isopropyl (compound 5) coupled with the 2-thienyl 14 Å internal cavity motif afforded a 6 to 33-fold improvement in potency for HDAC1 and 2 with modest selectivity versus HDAC3 (Table 2). This gain of potency combined with the minimal atomic size of this group maintained excellent ligand efficiency of 0.37. However, the tert-butyl compound 6 exceeded the steric limits of the channel and led to a significant loss in potency against HDAC1, 2 and 3. In contrast, the constrained cyclopropyl group (compound 7) afforded a dramatic 100-fold improvement in potency for HDAC2.
Increasing linker ring size (compounds 8–10) combined with hydrophobic (p-fluorophenyl or 2-thienyl) 14 Å internal cavity motifs increased potency on HDAC1 only, suggesting that larger hydrophobic and sp3-rich linkers are tolerated in HDAC2 but do not provide additional binding energy. Contrary to observations in the hydroxamic acid chemical series,32 sp2-hybridization α to the carbonyl in BRD2283 had minimal effect on the inhibitor activity towards Class I HDACs. The addition of hydrophilic linker groups gave mixed effects towards binding affinity. The basic N-methyl piperidine (compound 11) was not tolerated as its inhibitory activity on all HDACs suffered a ≥80-fold loss. The highly hydrophobic 11 Å channel, lined by two phenylalanines (Fig. 1), does not tolerate the hydrophilic piperidine ring which is protonated at physiological pH. In contrast, the neutral N-acetylpiperidine (BRD3349) or pyran (BRD4884) derivatives provided highly potent and selective HDAC1, 2 inhibitors (Table 2, BRD4884: IC50 0.029 μM and 0.062 μM on HDAC1 and HDAC2 respectively with ≥17-fold selectivity versus HDAC3). Interestingly, replacement of the p-fluorophenyl with a 4-pyridyl as an internal cavity motif (cf.BRD4884 to compound 12) reduced potency on HDAC1, 2 and 3 by 18 to 80-fold. In this carbamide series, the combination of sp3-rich linker with a 14 Å cavity hydrophobic aryl group is preferred, affording highly potent and selective HDAC1 and 2 inhibitors. Next, we evaluated the in vitro kinetic binding properties towards HDACs 1, 2 and 3, through progression curve analyses at various inhibitor concentrations and substrate conversion dilution experiments monitored continuously for 4 hours. Analysis of BRD4884 kinetic parameters revealed slow-on/slow-off kinetics for HDAC2, but a shift to fast-on/faster-off kinetics for HDAC1, leading to a 7-fold longer half-life on HDAC2 (T1/2 143 min) versus HDAC1 (T1/2 20 min; Table 2 and ESI Table 2†). This binding profile provides kinetic selectivity for HDAC2 and good thermodynamic selectivity for HDAC1, 2 versus HDAC3. Further characterization of BRD4884 in mice revealed good pharmacokinetic properties (T1/2 = 0.9 hours) including excellent brain permeability (brain-to-plasma ratio of 1.29 based on AUC, see ESI Fig. 1†) and a moderate predicted free fraction (6%) in brain based on a tissue binding assay.
Intrigued by the potency and kinetic selectivity towards HDAC2 of these sp3-rich carbamide-linked inhibitors, we investigated whether the hydrophobic 11 Å channel could tolerate alternative chemotypes such as carbamates and ureas (Table 3).
|
|
HDAC isoform inhibition IC50a (μM) | [Brain]/[plasma] and brain free fractionb (%) | ||||
|---|---|---|---|---|---|---|
| Compound | R1 group | R2 group | HDAC1 | HDAC2 | HDAC3 | |
| a Values are the mean of a minimum of two experiments. Data are shown as IC50 values in μM ± standard deviation. Compounds were tested in duplicate in a 12-point dose curve with 3-fold serial dilution starting from 33.33 μM. nd = not determined. b Brain free fraction estimated based on brain tissue binding experiments. | ||||||
| 13 |
|
|
0.611 ± 0.253 | 1.00 ± 0.34 | 2.60 ± 0.09 | nd |
| 14 |
|
|
0.069 ± 0.031 | 0.104 ± 0.028 | 0.861 ± 0.141 | nd |
| 15 |
|
|
0.071 ± 0.009 | 2.64 ± 1.13 | 13.05 ± 3.04 | nd |
| 16 |
|
|
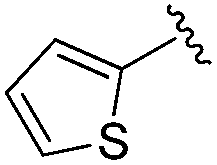
0.216 ± 0.050 | 0.912 ± 0.155 | 13.2 ± 2.19 | nd |
| 17 |
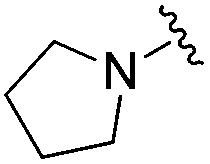
|
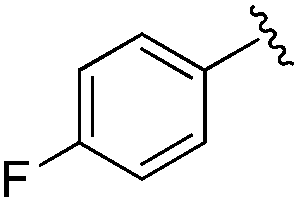
|
0.010 ± 0.002 | 0.059 ± 0.021 | 1.47 ± 0.25 | nd |
| BRD3321 |
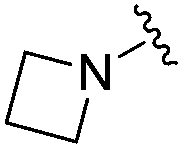
|
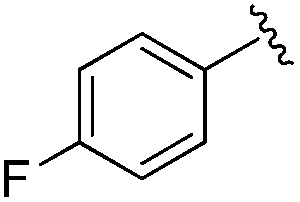
|
0.019 ± 0.005 | 0.233 ± 0.053 | 1.75 ± 0.25 | nd |
| BRD0302 |
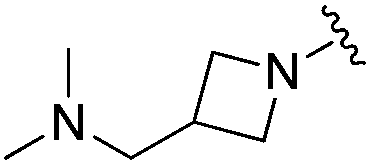
|
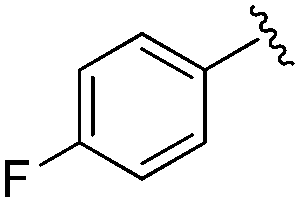
|
0.113 ± 0.015 | 1.29 ± 0.55 | 9.22 ± 2.06 | nd |
| Ki (μM) | ||||||
| 0.111 | 2.74 | 17.7 | ||||
| Residence time T 1/2 (min) | ||||||
| 308 | 375 | 231 | ||||
| BRD6688 |
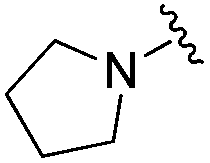
|

|
0.021 ± 0.013 | 0.100 ± 0.048 | 11.48 ± 2.54 | 0.26 |
| 54% | ||||||
| Residence time T 1/2 (min) | ||||||
| 65 | 381 | 280 | ||||
| 18 |
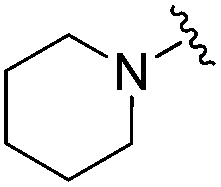
|
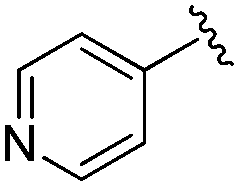
|
0.093 ± 0.022 | 0.176 ± 0.100 | 10.15 ± 3.27 | nd |
| 19 |
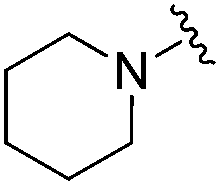
|
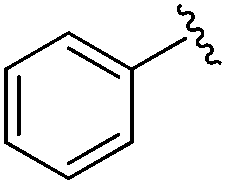
|
0.024 ± 0.001 | 0.271 ± 0.086 | 1.96 ± 1.34 | nd |
| BRD3227 |
|
|
0.043 ± 0.024 | 0.291 ± 0.141 | 23.5 ± 6.7 | 0.01 |
| 20 |
|
|
0.035 ± 0.012 | 0.238 ± 0.107 | 5.07 ± 0.93 | 0.19 |
| 21% | ||||||
| Residence time T 1/2 (min) | ||||||
| 165 | 513 | 495 | ||||
| BRD3386 |
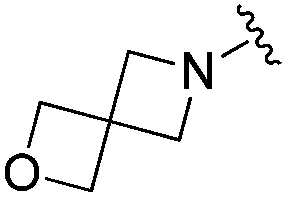
|
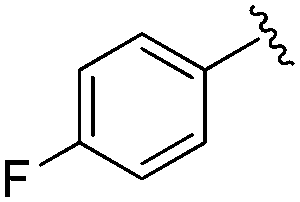
|
0.026 ± 0.007 | 0.178 ± 0.058 | 3.13 ± 0.90 | 0.34 |
| 22% | ||||||
| Residence time T 1/2 (min) | ||||||
| 570 | 660 | 495 | ||||
| BRD8951 |
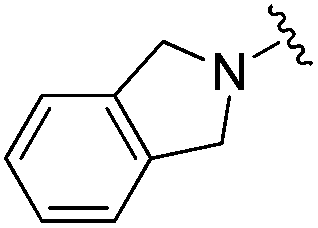
|
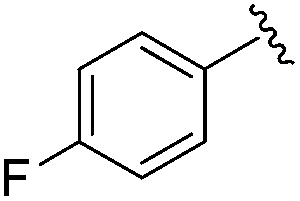
|
0.001 ± 0.001 | 0.011 ± 0.003 | 0.544 ± 0.205 | 0.27 |
| 2% | ||||||
| Residence time T 1/2 (min) | ||||||
| 2100 | 788 | ND | ||||
| BRD4161 |
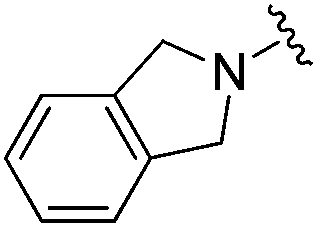
|
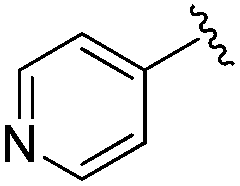
|
0.007 ± 0.002 | 0.045 ± 0.010 | 3.46 ± 0.89 | 0.11 |
| 23% | ||||||
| Residence time T 1/2 (min) | ||||||
| 430 | 788 | 770 | ||||

We anticipated that these alternate chemotypes would affect the electronic nature of the carbonyl moiety and negatively influence its' ability to effectively chelate zinc. To our surprise, propyl carbamate 13 was an effective inhibitor of HDAC1 and 2 with low micromolar potencies. While an extensive exploration of the SAR in the carbamate series did not lead to selective HDAC2 inhibitors, it did produce potent HDAC1, 2 inhibitors. Compound 14 represents the most potent and selective HDAC1, 2 inhibitor of the carbamate series (Table 3; IC50 0.069 and 0.104 μM on HDAC1 and 2 respectively with 8-fold selectivity versus HDAC3).
Next we turned our attention to the nitrogen ortholog of carbamate 13 to explore the influence of alternative heteroatoms at this position. Propyl urea 15 and the N-methylated analog 16 displayed low micromolar potencies for HDAC1 and 2. On the basis of our computational models defining the topology of the 11 Å channel (Fig. 1B) and the observed SAR in the carbamide series we next examined conformationally constrained ureas. Cyclizing the N–Me motif in compound 16 onto the terminal propyl carbon provided compound 17, which displayed excellent potency and selectivity for HDACs 1, 2 (Table 3; IC50 0.010 and 0.059 μM on HDAC1 and 2 respectively with 25-fold selectivity versus HDAC3). Interestingly the smaller azetidine linked ureas, BRD3321 and BRD0302, combined with a p-fluorophenyl 14 Å cavity motif demonstrated >12-fold selectivity for HDAC1 over HDAC2 (BRD0302 HDAC1 Ki = 0.111 μM vs. HDAC2 Ki = 2.74 μM, 25-fold selectivity, no kinetic selectivity was observed). These compounds represent some of the most thermodynamically selective HDAC1 inhibitors reported to date and reinforce the notion that differentiation between these two isoforms is possible. An important SAR distinction in the urea series versus the carbamide series is that heteroaromatic internal cavity motifs (cf.BRD6688vs. compound 17) retain potency towards HDAC1 and 2 (ligE = 0.33) allowing us to tune physicochemical properties through substitutions in this portion of the molecule. Additionally, the 4-pyridyl motif in BRD6688 provides increased selectivity for HDACs 1 and 2 (≥115-fold) reflected by an HDAC3 IC50 of 11.4 μM. More importantly, BRD6688 possesses preferential binding kinetics with extended half-life on HDAC2 compared to HDAC1 (381 min versus 65 min, 6-fold selectivity). To determine the optimal cyclic urea motif, we synthesized piperidine and morpholine analogs 18–20 and BRD3227 which led to a slight loss in potency on both HDAC1 and 2. In HDAC2, the apparent steric limit presented by a 6-membered linker group in the 11 Å channel and the corresponding loss in potency could not be compensated for by the use of a hydrophobic 14 Å cavity group (compound 19) and/or by ring substitutions (e.g. morpholine in compound 20 and 4-acetamide in BRD3227). Also, no significant increase in potency was observed when using the sterically less demanding oxa-aza-spiroheptane ring33 in BRD3386 as an alternative to the morpholine. In order to minimize sp3 steric components and capitalize on potential π–π interactions with Phe 155 and 210 which line the 11 Å channel (Fig. 1A and B), we synthesized isoindoline ring systems (BRD8951 and BRD4161). These compounds displayed improved potency on both HDAC1 and HDAC2 irrespective of the nature of the internal 14 Å cavity motif. Analysis of the isoform binding kinetics of these more potent HDAC1, 2 inhibitors showed no kinetic selectivity and presented no improvement relative to BRD6688.
We have identified and characterized the first kinetically selective HDAC2 inhibitors in two novel and distinct chemical series (full binding kinetics provided in ESI Table 2†). The carbamide BRD4884 and the urea BRD6688 possess selective binding kinetics for HDAC2 (T1/2 = 143 and 381 min respectively) compared to the highly homologous isoform HDAC1 (T1/2 = 20 and 65 min respectively). Interestingly, these kinetically selective HDAC2 ortho-aminoanilide based inhibitors rely on the incorporation of sp3-rich linker motifs coupled with aryl and/or heteroaryl 14 Å cavity motifs. Both compounds show excellent HDAC2 thermodynamic selectivity versus other Class I (>17-fold) and Class II (>500-fold) HDAC isoforms tested (ESI Table 1†). Moreover, BRD4884 and BRD6688 display good to excellent brain penetration (ESI Fig. 1†), low brain tissue binding, low potential cardiac toxicity, and high specificity versus a broad panel of biological targets (Tables 2 and 3 and ESI Table 3†). To better define HDAC isoform selectivity in brain we integrated the in vitro kinetic binding parameters, in vivo pharmacokinetic properties (including brain free fraction) and the HDAC enzyme concentration in brain34 by simulating target engagement profiles over time using numerical integrations over a system of differential equations describing the distribution of enzyme states (Fig. 2, see ESI for detailed description of method and input parameters†). Good correlation between in vitro and in vivo derived kinetic binding parameters for small molecule inhibitors of HDACs 1, 2 and 3 has been demonstrated using brain tissue autoradiography.34
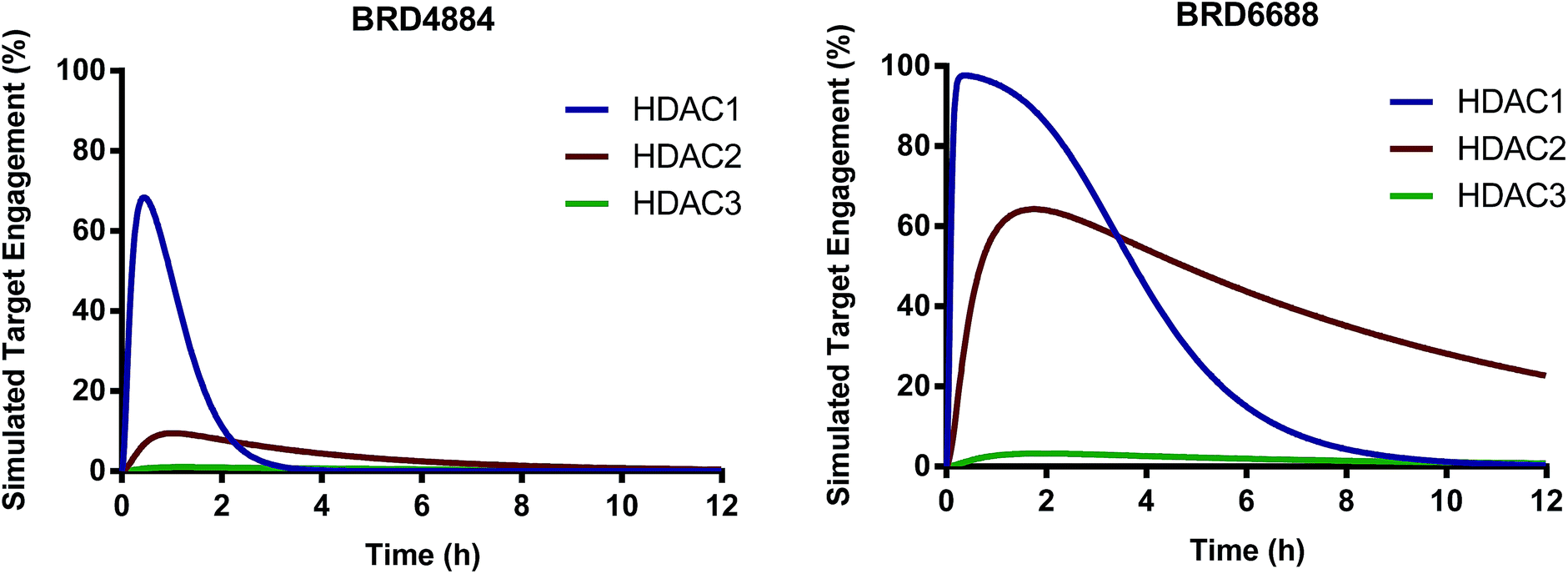 | ||
| Fig. 2 Simulated target engagement profiles for HDAC1, 2 and 3 in brain for BRD4884 and BRD6688 at 10 mg kg−1 dose. | ||
The simulated target engagement profiles for both compounds are characterized by three phases of kinetic selectivity (ESI Fig. 2†); an initial phase (t = 0–60 min) of good kinetic selectivity for HDAC1 (BRD4884, 3.5–30-fold, BRD6688, 2.5–20-fold), an intermediate crossover stage with equivalent target engagement levels for HDAC1 and 2, followed by a terminal phase (BRD4884, t > 3 h; BRD6688, t > 6 h) of high and sustained kinetic selectivity for HDAC2 (BRD4884, 20–1000X; BRD6688, 3–50-fold). Both compounds exhibit high kinetic selectivity against HDAC3 throughout the simulation. There is, however, a substantial difference between the two compounds in the magnitude of HDAC2 target engagement, which is driven by differences in the measured free fraction (BRD6688, fu = 0.54; BRD4884, fu = 0.06) and, to a lesser extent, the slower on and off-rate for BRD6688. BRD6688 attains greater than 50% HDAC2 engagement for several hours whereas BRD4884 achieves no more than 10% HDAC2 engagement. Taken together, these compounds represent the state of the art HDAC2 selective inhibitors to probe the function of HDAC2 in brain via small molecule modulation.
To further validate the activity of these compounds, we investigated whether the in vitro biochemical activities against human recombinant enzymes and their respective kinetic profiles translated to functional cell based assays by measuring histone acetylation changes.
H3K9 and H4K12 have been implicated as potential HDAC2 substrates in HDAC2 KO and OE transgenic mice.12 However, these histone loci display acetylation changes in response to non-selective HDAC2 inhibitor treatment35,36 demonstrating the non-specific nature of theses loci towards HDAC2. Primary mouse forebrain neuronal cultures were treated with BRD4884 and BRD6688 (10 μM for 24 h) and monitored for acetylation changes at H3K9 and H4K12 relative to the vehicle control (Fig. 3).
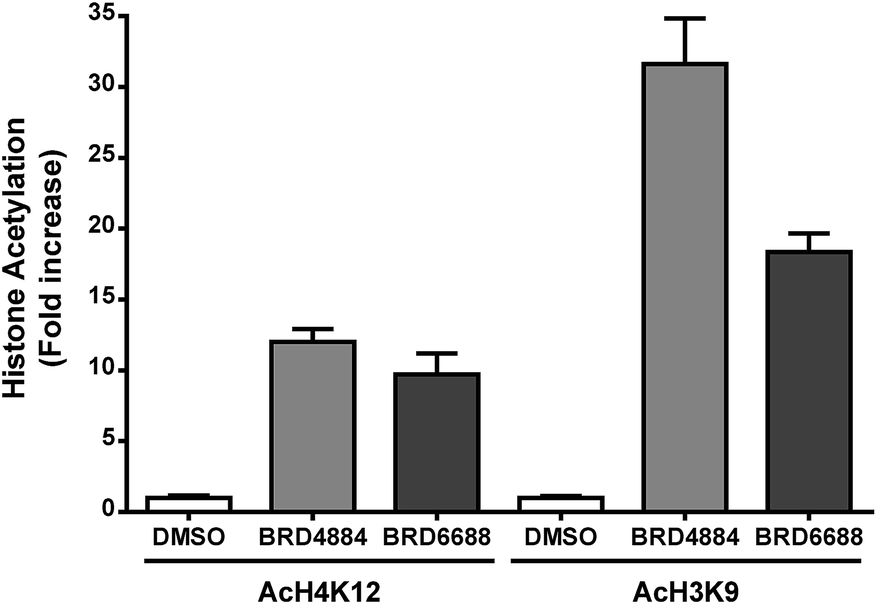 | ||
| Fig. 3 Increased acetylation of histones H4K12 and H3K9 in mouse forebrain primary neuronal cultures following treatment with kinetically selective HDAC2 inhibitors BRD4884 and BRD6688 (10 μM, 24 hours). Average of two experiments run in triplicates from separate dissections and cultures. | ||
Treatment with BRD4884 or BRD6688 produced significant increases in AcH4K12 and AcH3K9 confirming the inhibitory activity of these compounds towards endogenous HDACs. While these histone acetylation increases are indicative of HDAC inhibition, it is not clear whether these changes are driven solely through modulation of HDAC2 or through a combination of HDACs including HDACs 1 and 3. We speculate that the attenuated change in AcH3K9 demonstrated by BRD6688 (cf.BRD4884) is due in part to its' superior selectivity for HDAC2 relative to HDAC3, the most highly expressed HDAC isoform in the brain.37
To further characterize the translational potential of kinetically selective HDAC2 inhibitors in cognitive disorders, BRD4884 and BRD6688 were evaluated in CK-p25 mice, a murine model of neurodegeneration with profound deficits in spatial and associative memory.38,39 Overexpression of p25 protein is controlled by a doxycycline-repressed, calcium/calmodulin-dependent protein kinase II (CaMKII) promoter.38 Six week induction recapitulates many hallmark features of Alzheimer's disease, including progressive neuronal loss, tau pathology, β-amyloid accumulation, cognitive dysfunction and impaired synaptic plasticity.2,39,40 Daily treatment for 10 days with BRD4884 or BRD6688 (10 and 1‡ mg kg−1, i.p. dosing respectively, Fig. 4A), rescued the memory defects associated with p25 induced neurodegeneration in contextual fear conditioning, a hippocampal dependent form of learning (Fig. 4B). Remarkably, BRD6688 daily compound treatment at 1 mg kg−1 in p25 induced animals restored the freezing response to normal levels compared to the vehicle treated non-induced p25 littermates (red vs. white bar).
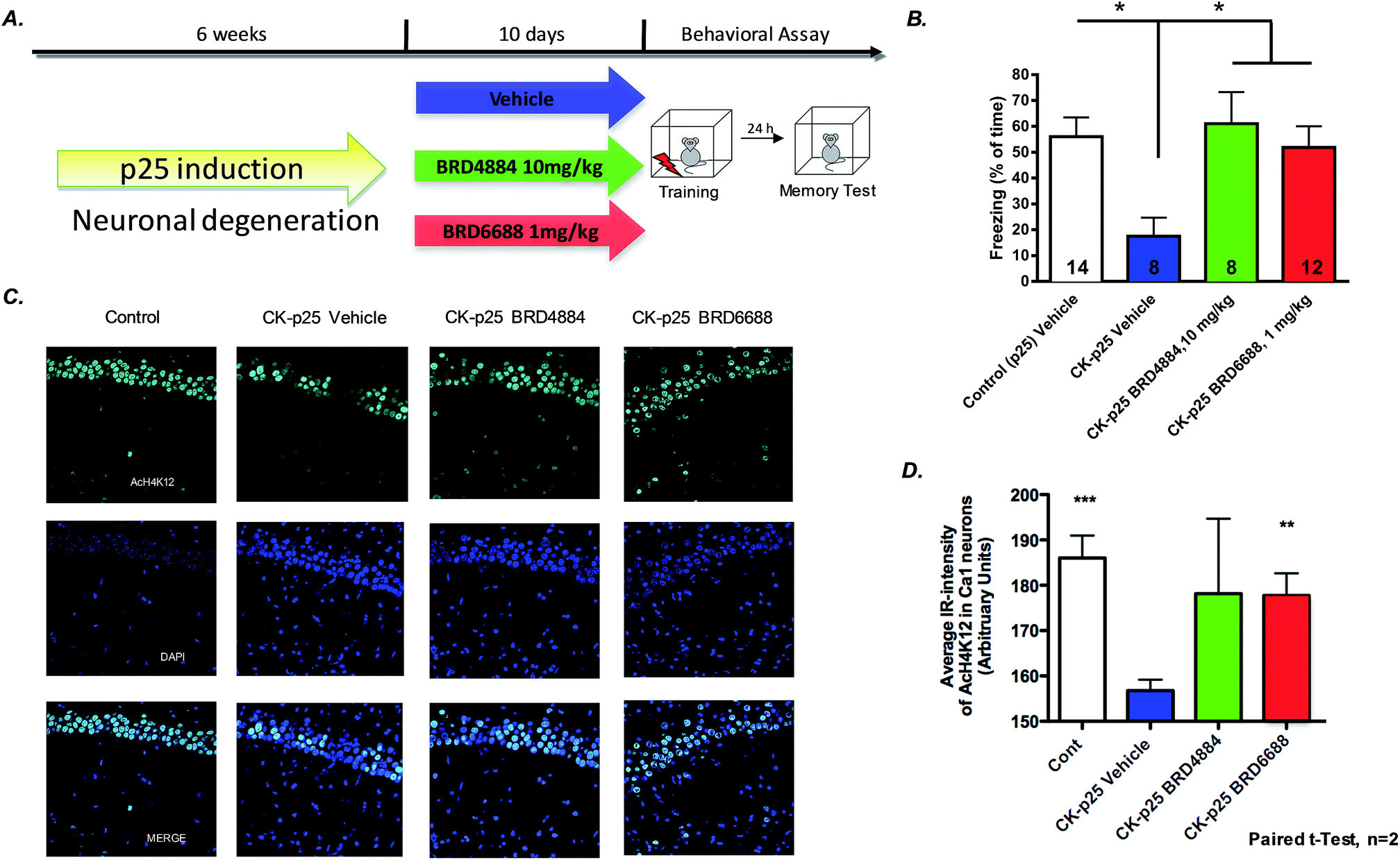 | ||
| Fig. 4 A. Study design for CK-p25 induced neurodegenerative model and testing in contextual fear conditioning paradigm. (B) BRD4884 and BRD6688 enhance freezing time in CK-p25 mice in a contextual fear conditioning behavioral paradigm. One-way ANOVA comparison followed by Dunnet's posthoc analysis; *p < 0.05; n is depicted in each bars. (C) Hippocampal sections from CK-p25 mice after 10 day treatment with BRD6688 demonstrate increased H4K12 acetylation (paired t-test). (D) Quantitation of increased acetylation in hippocampal slices after BRD6688 treatment. | ||
Furthermore, compound treatment corresponded with increased H4K12 acetylation in hippocampal CA1 neurons compared to the vehicle treated group (Fig. 3C and D, BRD6688 treatment effect was significant in paired t-test). Taken together, our results demonstrate that these novel and kinetically selective HDAC2 inhibitors engage HDACs in the brain and elicit acetylation changes at doses that produce enhanced learning behaviors in cognitively challenged mice.
Conclusions
Here we demonstrate for the first time that the selective inhibition of histone deacetylase 2 (HDAC2) (versus all other zinc dependent HDACs) is feasible. Starting with the ortho-aminoanilide chemical series and focusing on a linker-centric strategy, we developed highly optimized compounds suitable for CNS applications. Remarkably, we demonstrated that the binding kinetics of these inhibitors towards individual isoforms is tunable through a combination of linker and internal 14 Å cavity motifs. These structural combinations, exemplified by BRD4884 and BRD6688, demonstrate kinetic selectivity for HDAC2 vs. HDAC1, an isoform with 95% similarity within the catalytic binding domain. In addition, these kinetically selective HDAC2 inhibitors increased histone acetylation (H4K12 and H3K9) in primary mouse neuronal cultures as well as in hippocampal CA1 neurons in CK-p25 mice. The increased histone acetylation in brain serves as a surrogate pharmacodynamic marker of HDAC engagement and was consistent with our observed brain pharmacokinetic properties.We demonstrated that HDAC2 selective inhibitors rescue the cognitive deficits in CK-p25 mice, a model of neurodegeneration; in a Pavlovian fear conditioning behavioral assay. The cognitive improvements observed in these hippocampal-dependent memory processes recapitulate previous results reported by Guan et al. on the effects elicited via the genetic knockout of HDAC2,12 as well as through targeted RNA interference (RNAi)-mediated HDAC2 gene silencing selectively within the hippocampus.2 Our studies suggest that a sustained low level of HDAC2 engagement (∼10% for BRD4884) by an orthosteric kinetically biased small molecule inhibitor is sufficient for biological activity. While these compounds demonstrate sufficient selectivity versus HDAC3 to preclude its' role in the biological effects observed, these compounds, particularly BRD4884, do not achieve sufficient selectivity versus HDAC1. It is possible that the intermittent inhibition of HDAC1 may play a role in the effects observed. Clearly experimental efforts are needed to confirm these target engagement profiles in vivo. Taken together, our studies suggest that the pharmacological inhibition of HDAC2 may enhance learning and memory and potentially rescue the observed cognitive deficits in multiple neuro-psychiatric disorders such as schizophrenia and PTSD. Additionally, isoform selective inhibitors may mitigate some of the known mechanism-based toxicological effects associated with the inhibition of multiple HDACs, particularly the concominant inhibition of HDAC1 and 2.41 Also, future studies will determine the potency and selectivity of this class of HDAC inhibitors towards distinct Class I HDAC complexes that are known to exist in the brain and play different biological functions.12
Our studies open the way for the design of highly ligand efficient and selective small molecule HDAC inhibitors optimized for central nervous system disorders. In AD, while drugs targeting the clearance of β-amyloid have failed to slow disease progression and improve cognitive measures; combination therapy with HDAC2 selective inhibitors could potentially restart synaptic function and memory formation. These novel small molecule inhibitors can be used as tools for probing the biological functions and relevance of the different HDAC isoforms and will catalyze the evaluation of their therapeutic potential in treating neurological disorders.
Experimental
Synthetic procedures
Detailed synthetic procedures are described in ESI.†Materials and methods
Funding sources
This research was funded by the Stanley Medical Research Institute, the JPB Foundation (L.H.T.) and the NIH/NIDA (S.J.H., R01DA028301)Conflict of interest disclosure
L.H.T., S.J.H, and E.B.H are consultants to Rodin Therapeutics which has licensed compounds from the Broad Institute.Acknowledgements
We would like to thank Dr Steve Johnston for analytical/purification support and Nhien Le for compound management support.Notes and references
- J. Gräff and L. H. Tsai, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 311–330 CrossRef PubMed.
- J. Gräff, D. Rei, J. S. Guan, W. Y. Wang, J. Seo, K. M. Hennig, T. J. Nieland, D. M. Fass, P. F. Kao, M. Kahn, S. C. Su, A. Samiei, N. Joseph, S. J. Haggarty, I. Delalle and L. H. Tsai, Nature, 2012, 483, 222–226 CrossRef PubMed.
- M. Weiwer, M. C. Lewis, F. F. Wagner and E. B. Holson, Future Med. Chem., 2013, 5, 1491–1508 CrossRef CAS PubMed.
- J. Gräff, N. F. Joseph, M. E. Horn, A. Samiei, J. Meng, J. Seo, D. Rei, A. W. Bero, T. X. Phan, F. Wagner, E. Holson, J. Xu, J. Sun, R. L. Neve, R. H. Mach, S. J. Haggarty and L. H. Tsai, Cell, 2014, 156, 261–276 CrossRef PubMed.
- T. Abel and R. S. Zukin, Curr. Opin. Pharmacol., 2008, 8, 57–64 CrossRef CAS PubMed.
- M. Mahgoub and L. M. Monteggia, Neurotherapeutics, 2013, 10, 734–741 CrossRef CAS PubMed.
- D. M. Fass, M. M. Kemp, F. A. Schroeder, F. F. Wagner, Q. Wang and E. B. Holson, Histone Acetylation and Deacetylation., Weinheim, 2012 Search PubMed.
- M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- J. M. Alarcon, G. Malleret, K. Touzani, S. Vronskaya, S. Ishii, E. R. Kandel and A. Barco, Neuron, 2004, 42, 947–959 CrossRef CAS PubMed.
- N. Govindarajan, P. Rao, S. Burkhardt, F. Sananbenesi, O. M. Schluter, F. Bradke, J. Lu and A. Fischer, EMBO Mol. Med., 2013, 5, 52–63 CrossRef CAS PubMed.
- J. S. Guan, S. J. Haggarty, E. Giacometti, J. H. Dannenberg, N. Joseph, J. Gao, T. J. Nieland, Y. Zhou, X. Wang, R. Mazitschek, J. E. Bradner, R. A. DePinho, R. Jaenisch and L. H. Tsai, Nature, 2009, 459, 55–60 CrossRef CAS PubMed.
- G. Li, H. Jiang, M. Chang, H. Xie and L. Hu, J. Neurol. Sci., 2011, 304, 1–8 CrossRef CAS PubMed.
- M. Malvaez, S. C. McQuown, G. A. Rogge, M. Astarabadi, V. Jacques, S. Carreiro, J. R. Rusche and M. A. Wood, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2647–2652 CrossRef CAS PubMed.
- S. C. McQuown and M. A. Wood, Curr. Psychiatr. Rep., 2010, 12, 145–153 CrossRef PubMed.
- M. J. Morris, M. Mahgoub, E. S. Na, H. Pranav and L. M. Monteggia, J. Neurosci., 2013, 33, 6401–6411 CrossRef CAS PubMed.
- F. F. Wagner, M. Weiwer, M. C. Lewis and E. B. Holson, Neurotherapeutics, 2013, 10, 589–604 CrossRef CAS PubMed.
- D. Kim, C. L. Frank, M. M. Dobbin, R. K. Tsunemoto, W. Tu, P. L. Peng, J. S. Guan, B. H. Lee, L. Y. Moy, P. Giusti, N. Broodie, R. Mazitschek, I. Delalle, S. J. Haggarty, R. L. Neve, Y. Lu and L. H. Tsai, Neuron, 2008, 60, 803–817 CrossRef CAS PubMed.
- R. H. Wilting, E. Yanover, M. R. Heideman, H. Jacobs, J. Horner, J. van der Torre, R. A. DePinho and J. H. Dannenberg, EMBO J., 2010, 29, 2586–2597 CrossRef CAS PubMed.
- R. A. Copeland, D. L. Pompliano and T. D. Meek, Nat. Rev. Drug Discovery, 2006, 5, 730–739 CrossRef CAS PubMed.
- J. C. Bressi, A. J. Jennings, R. Skene, Y. Wu, R. Melkus, R. De Jong, S. O'Connell, C. E. Grimshaw, M. Navre and A. R. Gangloff, Bioorg. Med. Chem. Lett., 2010, 20, 3142–3145 CrossRef CAS PubMed.
- J. L. Methot, P. K. Chakravarty, M. Chenard, J. Close, J. C. Cruz, W. K. Dahlberg, J. Fleming, C. L. Hamblett, J. E. Hamill, P. Harrington, A. Harsch, R. Heidebrecht, B. Hughes, J. Jung, C. M. Kenific, A. M. Kral, P. T. Meinke, R. E. Middleton, N. Ozerova, D. L. Sloman, M. G. Stanton, A. A. Szewczak, S. Tyagarajan, D. J. Witter, J. P. Secrist and T. A. Miller, Bioorg. Med. Chem. Lett., 2008, 18, 973–978 CrossRef CAS PubMed.
- O. M. Moradei, T. C. Mallais, S. Frechette, I. Paquin, P. E. Tessier, S. M. Leit, M. Fournel, C. Bonfils, M. C. Trachy-Bourget, J. Liu, T. P. Yan, A. H. Lu, J. Rahil, J. Wang, S. Lefebvre, Z. Li, A. F. Vaisburg and J. M. Besterman, J. Med. Chem., 2007, 50, 5543–5546 CrossRef CAS PubMed.
- C. J. Chou, D. Herman and J. M. Gottesfeld, J. Biol. Chem., 2008, 283, 35402–35409 CrossRef CAS PubMed.
- A. M. Kral, N. Ozerova, J. Close, J. Jung, M. Chenard, J. Fleming, B. B. Haines, P. Harrington, J. Maclean, T. A. Miller, P. Secrist, H. Wang and R. W. Heidebrecht Jr, Biochemistry, 2014, 53, 725–734 CrossRef CAS PubMed.
- B. E. Lauffer, R. Mintzer, R. Fong, S. Mukund, C. Tam, I. Zilberleyb, B. Flicke, A. Ritscher, G. Fedorowicz, R. Vallero, D. F. Ortwine, J. Gunzner, Z. Modrusan, L. Neumann, C. M. Koth, P. J. Lupardus, J. S. Kaminker, C. E. Heise and P. Steiner, J. Biol. Chem., 2013, 288, 26926–26943 CrossRef CAS PubMed.
- J. L. Methot, D. M. Hoffman, D. J. Witter, M. G. Stanton, P. Harrington, C. Hamblett, P. Siliphaivanh, K. Wilson, J. Hubbs, R. Heidebrecht, A. M. Kral, N. Ozerova, J. C. Fleming, H. Wang, A. A. Szewczak, R. E. Middleton, B. Hughes, J. C. Cruz, B. B. Haines, M. Chenard, C. M. Kenific, A. Harsch, J. P. Secrist and T. A. Miller, ACS Med. Chem. Lett., 2014, 5, 340–345 CrossRef CAS PubMed.
- Y. J. Seo, Y. Kang, L. Muench, A. Reid, S. Caesar, L. Jean, F. Wagner, E. Holson, S. J. Haggarty, P. Weiss, P. King, P. Carter, N. D. Volkow, J. S. Fowler, J. M. Hooker and S. W. Kim, ACS Chem. Neurosci., 2014, 5, 588–596 CrossRef CAS PubMed.
- L. Riva, S. M. Blaney, R. Dauser, J. G. Nuchtern, J. Durfee, L. McGuffey and S. L. Berg, Clin. Cancer Res., 2000, 6, 994–997 CAS.
- Y.-L. Zhang, E. Holson and F. F. Wagner, WO 2013067391, 2013.
- M. Rai, E. Soragni, C. J. Chou, G. Barnes, S. Jones, J. R. Rusche, J. M. Gottesfeld and M. Pandolfo, PLoS One, 2010, 5, e8825 Search PubMed.
- F. F. Wagner, D. E. Olson, J. P. Gale, T. Kaya, M. Weiwer, N. Aidoud, M. Thomas, E. L. Davoine, B. C. Lemercier, Y. L. Zhang and E. B. Holson, J. Med. Chem., 2013, 56, 1772–1776 CrossRef CAS PubMed.
- J. A. Burkhard, C. Guerot, H. Knust and E. M. Carreira, Org. Lett., 2012, 14, 66–69 CrossRef CAS PubMed.
- Y. Wang, Y. L. Zhang, K. Hennig, J. P. Gale, Y. Hong, A. Cha, M. Riley, F. Wagner, S. J. Haggarty, E. Holson and J. Hooker, Epigenetics, 2013, 8, 756–764 CrossRef CAS PubMed.
- D. M. Fass, S. A. Reis, B. Ghosh, K. M. Hennig, N. F. Joseph, W. N. Zhao, T. J. Nieland, J. S. Guan, C. E. Kuhnle, W. Tang, D. D. Barker, R. Mazitschek, S. L. Schreiber, L. H. Tsai and S. J. Haggarty, Neuropharmacology, 2013, 64, 81–96 CrossRef CAS PubMed.
- M. Naldi, N. Calonghi, L. Masotti, C. Parolin, S. Valente, A. Mai and V. Andrisano, Proteomics, 2009, 9, 5437–5445 CrossRef CAS PubMed.
- R. S. Broide, J. M. Redwine, N. Aftahi, W. Young, F. E. Bloom and C. J. Winrow, J. Mol. Neurosci., 2007, 31, 47–58 CrossRef CAS.
- J. C. Cruz, H. C. Tseng, J. A. Goldman, H. Shih and L. H. Tsai, Neuron, 2003, 40, 471–483 CrossRef CAS.
- A. Fischer, F. Sananbenesi, P. T. Pang, B. Lu and L. H. Tsai, Neuron, 2005, 48, 825–838 CrossRef CAS PubMed.
- P. Giusti-Rodriguez, J. Gao, J. Gräff, D. Rei, T. Soda and L. H. Tsai, J. Neurosci., 2011, 31, 15751–15756 CrossRef CAS PubMed.
- O. Bruserud, C. Stapnes, E. Ersvaer, B. T. Gjertsen and A. Ryningen, Curr. Pharm. Biotechnol., 2007, 8, 388–400 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Compound synthesis and characterization; 1HNMR spectra, HPLC or UPLC spectral traces; HDAC enzymatic assay protocol; IC50s for representative compounds for HDACs 1–9; full kinetic parameters for BRD4884 and BRD6688; progression and dissociation curves for BRD6688; pharmacokinetic graphs and parameters for representative compounds; kinetic selectivity profiles for BRD4884 and BRD6688; in vitro pharmacology, pharmacokinetic protocols; target engagement simulation protocol; molecular modelling and docking protocols; neuronal cell based assay protocol, behavioural studies protocols. See DOI: 10.1039/c4sc02130d |
| ‡ 1 mg kg−1 dose of BRD6688 was chosen due to tolerability issues observed at 10 mg kg−1: mortality in 5 out of 10 mice over 10 day treatment due to unknown cause in a single study. No toxicity was observed CK-p25 mice treated at 1 mg kg−1 or in wild-type male C57BL/6 mice treated at 30 mg kg−1 daily for 10 consecutive days. |
| This journal is © The Royal Society of Chemistry 2015 |