
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of storage conditions and storage time on silver nanoparticles' physicochemical properties and implications for their biological effects†
E.
Izak-Nau‡
ab,
A.
Huk‡
bc,
B.
Reidy‡
d,
H.
Uggerud
e,
M.
Vadset
e,
S.
Eiden
a,
M.
Voetz
a,
M.
Himly
b,
A.
Duschl
b,
M.
Dusinska
c and
I.
Lynch
*f
aBayer Technology Services GmbH, Leverkusen, Germany
bDepartment of Molecular Biology, University of Salzburg, Salzburg, Austria
cHealth Effects Laboratory, Department of Environmental Chemistry, NILU-Norwegian Institute for Air Research, Kjeller, Norway
dCentre for BioNano Interactions, School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland
eInorganic Group, Department of Environmental Chemistry, NILU-Norwegian Institute for Air Research, Kjeller, Norway
fSchool of Geography, Earth & Environmental Sciences, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: i.lynch@bham.ac.uk
First published on 25th September 2015
Abstract
It is increasingly recognized that nanoparticles (NPs) can ‘age’ while stored, and that the impact of this may lead to divergent results in terms of the observed toxicity of nominally the same NPs. The main goal of this study was to investigate whether (and to what extent) changes in silver (Ag) NPs' properties occur over time and whether storage of the dispersions under different conditions impacts their stability and ageing mechanism, as a function of the NPs' surface capping/charge. We found that both storage time/conditions and surface chemistry of AgNPs influenced the evolution of the NP properties over time, and that the resulting changes in the NPs' physicochemical properties influenced their toxicity. Observed changes in Ag NPs' toxicity were related to different processes such as NP agglomeration, dissolution, oxidation, capping agent degradation as well attachment of Ag+ ions to container walls. Thus, NP ‘aging’ effects as described here can be a significant contributor to the contradictory toxicity results observed in the literature for identical NPs, and NP ageing should thus be assessed in parallel with toxicity assessment as best practice.
Introduction
Nanoparticles (NPs) possess outstanding properties, which make them applicable in many areas of our daily life. Among the various NPs which are used commercially, silver NPs (AgNPs) are found in a wide number of consumer products ranging from food packaging or clothes to medical devices and implants.1–4 AgNPs have attracted attention because of their specific characteristics including good conductivity, catalytic activity, and antibacterial character.5–8 Despite the range of Ag NP-containing goods being introduced onto the market, there is a considerable lack of knowledge concerning their effects on consumers and the environment. Many studies evaluating the toxicity of AgNPs in vitro and in vivo have been published, however, the conclusions are inconsistent and it is not yet entirely clear whether the toxicity is primarily related to dissolved ionic forms of silver (Ag+) or also results from the NP forms (Ag0 NPs).9–11 This can be correlated with a lack of sufficient characterization of the materials being studied, including their speciation between Ag+ ions and Ag0 NPs and other insoluble forms, especially at the time of toxicological evaluation.Many NPs are likely to change their properties while stored under different conditions, and may also ‘age’ or evolve with time even under optimal storage conditions.12–14 Such temporal changes in NPs' properties may modulate their toxicity, by increasing or decreasing their dissolution rate, or altering their agglomeration state which can affect bioavailability, leading to discrepancies of administered vs. delivered dose.13–16 It is likely that NPs' stability depends not only on the storage time and conditions but also on the capping agent used to stabilize them.13,17–19 Published studies show that a wide variety of capping agents have been used to stabilize AgNPs against agglomeration and/or dissolution, with the most common being sodium citrate and organic polymers such as poly(ethylene glycol) or polyvinylpyrrolidone (PVP).20–22 The stability of AgNPs has been previously investigated,23,24 indicating that their stability strongly influences their toxicity.24–27
The technical challenges in the assessment of NP toxicity have long been recognized in the field and a recent meta-analysis of the nanotoxicological literature has highlighted the need to get all aspects of the relevant experiments under control.28 The present study aims to contribute to this effort by investigating one specific topic, particle aging, in the case of AgNPs as a function of storage conditions and capping agent composition and charge. While some reports describing the time-dependent behaviour of AgNPs have already been published,24,29 our research is unique in terms of the variety of stabilizing agents considered and the connection with how storage time/conditions can modulate toxicity impacts. The selected AgNPs are stabilized not only with the common capping agents, but also with novel chemicals that exhibit different surface charges. The differently stabilized/charged AgNPs (5–10 nm) were prepared via the same synthesis route and varied in terms of their surface chemistry by the use of different capping agents (negative charge: trisodium citrate or SDS; positive charge: chitosan or BYK-9076®; neutral: Tween 80® or DISPERBYK-192®). The use of two different capping agents for each charge used allows further comparison of the role of charge in stabilizing AgNPs (Table S1†).
The Ag NP's properties were examined immediately post synthesis (T0) and after 1 month (T1), 3 months (T3) and 6 months (T6) of storage under the following conditions: at room temperature in the presence (RT-DL) or absence of light (RT-D), and at lower temperature (in a fridge) in the absence of light (4 °C-D). The various dispersions were analysed by a wide range of physico-chemical approaches in order to assess temporal changes in the NPs' physicochemical properties during storage. The cytotoxicity of the AgNPs with different capping agents stored under the different conditions was studied using A549 cells at time points T0 and T6 by two independent laboratories using a panel of different end-points. High content screening (HCS) was used to assess multiple markers of cytotoxicity in parallel (Laboratory 1): cell count, mitochondrial potential, acidification of lysosomes and cell membrane permeabilisation. Additionally, toxicity of the AgNPs was measured using two standard cytotoxicity tests in accordance with good laboratory practice: plating efficiency (PE) to assess the colony forming ability, and relative growth activity (RGA) to measure cell proliferation and cell death (Laboratory 2, GLP-certified Health Effect Laboratory, NILU). Each laboratory performed the tests independently, according to their own established protocols.
Methods
Nanoparticle preparation
AgNPs were synthesized using a chemical reduction method (full protocol in the ESI†). Silver nitrate (AgNO3; Heraeus, Hanau, Germany) was used as the metal precursor and sodium borohydride (NaBH4; ACROS Organics, Geel, Belgium) as the reducing agent (modified protocol of Creighton et al.).30,31 Six different stabilizers were utilized: two negatively charged: 3-sodium citrate (Na3C6H5O7; Fisher Scientific, Schwerte, Germany) and sodium dodecyl sulfate (SDS; Sigma-Aldrich, St. Louis, MO, USA); two positively charged: chitosan (high molecular weight; Sigma-Aldrich, Taufkirchen, Germany) and BYK-9076® (BYK-Chemie, Wesel, Germany); and two neutral: Tween 80® (Sigma-Aldrich, Taufkirchen, Germany) and DISPERBYK-192® (BYK-Chemie, Wesel, Germany). These were selected for their stabilization ability and charge rather than with consideration of their potential toxicity.Nanoparticle characterization
The hydrodynamic size/size distribution and the zeta potentials of all AgNPs preparations were determined using a 90Plus particle size analyser (DLS; Brookhaven Instruments Corporation, Holtsville, NY).The primary NPs' size and shape were determined using a Phillips CM20 transmission electron microscope (TEM) working at 200 keV.
The absorbance of the AgNPs' dispersions was analyzed using an ultraviolet-visible spectrophotometer (UV-vis; Varian Cary 50).
The chemical composition of the NPs and the presence of different stabilizing agents were determined by PHI VersaProbe 5000 scanning X-ray photoelectron spectroscopy (XPS; Physical Electronics, Chanhassen, MN) immediately following synthesis.
The crystallite size and crystalline phase were obtained using an X-ray diffractometer (XRD) PANalytical EMPYREAN PIXcel with 3D Counter (PANalytical B.V., ALMELO, the Netherlands).
The equipment specifications and full experimental conditions for all NP characterization methods used are described in detail in the ESI.†
Nanoparticle stability
In order to study the effect of storage conditions on Ag NP stability, each of the prepared solutions (50 mL; 100 μg mL−1) was divided into three glass bottles (15 mL each); two were covered with aluminum foil and stored at RT (RT-D) and in a fridge (4 °C-D); one sample was kept at RT without foil and thus exposed to daylight (RT-DL). UV-vis, DLS, TEM, and zeta potential measurements were performed immediately post synthesis and subsequently after 1, 3 and 6 months of storage.The behaviour of all batches of NPs in cell culture medium (cMEM) was analysed using differential centrifugal sedimentation (DSC) after 6 months of storage (full details in the ESI†). DCS measurements were performed on CPS Disc Centrifuge DC24000 (CPS Instruments Europe, Oosterhout, The Netherlands).
Analysis of silver ions
Inductively coupled plasma mass spectrometry (ICP-MS; Agilent Technologies 7700x Series, Wilmington, USA) was applied to quantify the total Ag in the stock suspensions and to assess Ag+ release from the differently coated NPs during storage of the AgNPs for 6 months under the different conditions (4 °C-D, RT-D, RT-DL). Full method details are given in the ESI.†Cytotoxicity evaluation of AgNPs
Cell culture. Human lung carcinoma epithelial cells (A549) obtained from ATCC (item number CCL-185) were cultured in minimal essential medium (MEM; Gibco, USA), supplemented with 10% activated foetal calf serum (FCS) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, USA), hereafter called ‘complete MEM’ or cMEM; in a humidified atmosphere of 5% CO2 at 37 °C.
High content screening. All cytotoxicity measurements were performed on a Cellomics ArrayScan© VTI High Content Screening (HCS) Reader (ThermoScientific, Waltham, USA). Cells were seeded on 96-well plates (cell density: 5 × 103 cells per well, seeding volume 100 μL) and allowed to grow for 24 h. After this time, cells were exposed to the variously capped AgNPs or the appropriate dispersants/solvents for 24 h. AgNP suspensions or dispersants/solvents were prepared immediately before exposure to the cells (50 μL of suspension was added on top of the 100 μL medium already present in each well). AgNPs were added at concentrations varying from 0 to 8.25 μg mL−1. Fifty μL of fresh medium was added to control wells. In the last hour of the 24 h exposure time the following fluorescent dyes were added to each well: Hoechst/trihydrochloride/trihydrate (Hoechst 33342; 400 nM; Molecular Probes®, Waltham, USA), tetramethylrhodamine/methyl ester/perchlorate (TMRM; 20 nM; Molecular Probes®), LysoTracker® Green DND-26 (50 nM; Molecular Probes®), TO-PRO®-3 (800 nM; Molecular Probes®).
Triton™ X-100 (0.5 μL per well) was used as the positive control. Subsequently, the plates were scanned under conditions appropriate for living cells (37 °C, 5% CO2). Images were acquired using a 20× objective. The acquisition of 10 fields per well meant that ∼500 cells were analysed in each well (omitting the decrease of cell count due to NPs/solvent impact). Acquired images were analysed using the Cell Health Profiling BioApplication V4, allowing the simultaneous assessment of several cell health parameters (cell count, mitochondrial (membrane) potential, lysosomal acidification and membrane permeabilisation). Data obtained from the Cellomics BioApplication were further analysed using GraphPad Prism 5.0.
Cell culture. A549 cells were cultured in flasks in Roswell park memorial institute media 1640 (RPMI 1640, Sigma-Aldrich) with 10% heat-inactivated foetal bovine serum (FBS; 20 min at 55 °C; Sigma-Aldrich), 1% penicillin/streptomycin (Sigma-Aldrich) in a humidified atmosphere of 5% CO2 and 37 °C.
Relative growth activity (RGA). Cells were seeded in 6 well plates (1.6 × 105 cells per well) and grown for 24 h in a humidified atmosphere. Cells were then exposed to AgNPs for 24 h at 5 concentrations ranging from 1 to 10 μg mL−1, where 10 μg mL−1 was the maximum test concentration possible for these Ag NP stocks whereby the medium dilution did not compromise the cell viability and equates to a dilution of the particles 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with medium. After exposure, medium was removed; cells were washed, trypsinized and re-suspended in 1 mL of fresh medium. Ten μL of cell suspension was mixed with 10 μL of 0.4% trypan blue (Invitrogen) and the number of cells was measured, using a Countess™ Automated Cell Counter (Invitrogen). RGA was determined as the percentage increase in cell number between 0 and 24 h.
:10 with medium. After exposure, medium was removed; cells were washed, trypsinized and re-suspended in 1 mL of fresh medium. Ten μL of cell suspension was mixed with 10 μL of 0.4% trypan blue (Invitrogen) and the number of cells was measured, using a Countess™ Automated Cell Counter (Invitrogen). RGA was determined as the percentage increase in cell number between 0 and 24 h.
Potential interference of AgNPs with the Countess™ Automated Cell counter (Invitrogen, Norway) was studied in a cell free system, and no interference was observed.
Plating efficiency (PE). Cells were exposed to AgNPs as described above. After the exposure, NPs were removed; cells were washed, trypsinized, re-suspended in fresh medium and counted. One hundred cells per well were seeded in 6 well plates (one plate for each NP concentration) and left in an incubator for 10 days to grow and form colonies. After 10 days, colonies were stained with 1% methylene blue (Sigma-Aldrich) and counted manually. PE was calculated as the number of colonies relative to the number of colonies in the untreated control.
Statistical analysis
Data are expressed as mean ± SEM (standard error of the mean, toxicology evaluation) or mean ± SD (standard deviation, characterization) of three independent experiments. Differences between storage conditions were calculated by one-way analysis of variance (ANOVA) and post hoc Tukey tests were performed to evaluate the significance levels. To estimate IC50 values, a nonlinear regression analysis was used to fit four parametric logistic curves. The software used includes: Graph Pad Prism software, Microsoft® Excel added-Daniels XL toolbox.Results
Synthesis and characterization of AgNPs
Highly concentrated (100 μg mL−1) AgNPs were produced with the desired size (5–10 nm) and were thoroughly characterized (Table 1). The zeta potential results showed the expected surface charges (as per the applied capping agents), and were in the range considered to result in stable dispersions (for the positive and negative capping agents-typically considered to be ±30 mV). DLS data confirmed hydrodynamic sizes of 5–10 nm for all NPs, with the exception of Ag_Chitosan, for which particle size measurements were not possible as the size exceeded 1 μm at all time-points (most probably due to micelle formation of this agent). The TEM images showed a quasi-spherical shape of AgNPs and were indicative of good monodispersity (Fig. 1). The presence of the respective capping agents was confirmed by XPS. The XRD measurements proved the crystal nature of all AgNPs and indicated the crystal sizes.| Name | Ag_Citrate | Ag_SDS | Ag_Disperbyk | Ag_Tween | Ag_Byk | Ag_Chitosan |
|---|---|---|---|---|---|---|
| Surface charge [mV] | −42 ± 2 | −33 ± 1 | −3 ± 4 | −1 ± 2 | +27 ± 2 | +52 ± 2 |
| Concentration [μg mL−1] | 100 | |||||
| Shape | Spherical | |||||
| Crystal structure | Cubic | |||||
| Size [nm] | DLS: 5 ± 1 | DLS: 5 ± 1 | DLS: 9 ± 1 | DLS: 8 ± 1 | DLS: 9 ± 1 | DLS: — |
| TEM: 6 ± 2 | TEM: 6 ± 3 | TEM: 7 ± 3 | TEM: 6 ± 2 | TEM: 10 ± 3 | TEM: 9.8 ± 2.1 | |
| XRD: 6 ± 2 | XRD: 7 ± 2 | XRD: 6 ± 2 | XRD: — | XRD: 7 ± 3 | XRD: 7.1 ± 3.1 | |
| Surface chemistry [atom%] | C 44.6; O 41.6; Na 9.2; N 3.0; Ag 1.7 | C 48.1; O 40.2; Na 5.9; Ag 3.6; N 2.0; S 0.2 | C 72.8; O 24.5; Ag 1.6; N 1.0 | C 71.5; O 25.7; Na 2.6; Ag 0.2 | C 69.5; O 16.3; N 9.4; Ag 4.7 | C 46.3; O 41.3; N 7.3; Na 3.4; Ag 1.7 |
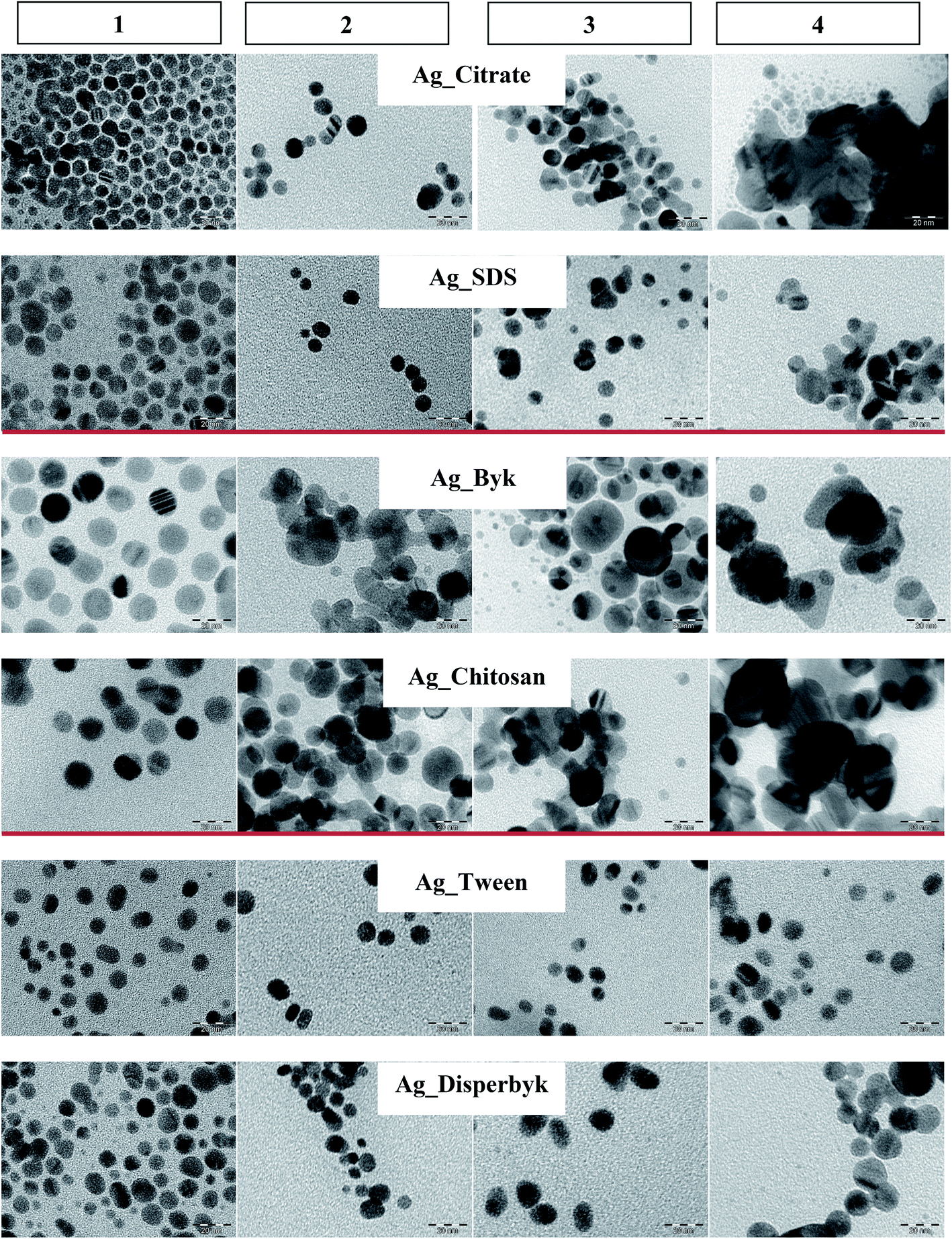 | ||
| Fig. 1 TEM pictures of AgNPs stored at: (1) T0, (2) T6:4 °C-D, (3) T6:RT-D, (4) T6:RT-DL (scale bar 20 nm). | ||
Changes in size and agglomeration during storage
Fig. 1 presents TEM images at T6 under the different storage conditions for the different capping agents. The negatively and positively charged NPs agglomerated while kept at RT-DL. In the case of the positively charged NPs, the particle size also increased at 4 °C-D and at RT-D where some agglomeration was observed. The neutral NPs displayed the best stability. However, in the case of the Ag_Disperbyk at RT-DL the size increased slightly by month 6.According to the DLS data (Fig. 2, Table S2†) the most significant changes in the NPs' sizes occurred at T6:RT-DL. At T1 there were no changes detectable by DLS in any case (irrespective of surface capping or storage conditions). In the case of the negatively charged NPs (Ag_Citrate and Ag_SDS), there was no significant change observed in the solutions stored at 4 °C-D and RT-D at any time. The Ag_Citrate seemed to be partially dissolved at T3:RT-DL, and at T6:RT-DL they were highly agglomerated. In the case of the Ag_SDS, the NPs size increased slightly at T3:RT-DL, and at T6:RT-DL this dispersion was highly agglomerated.
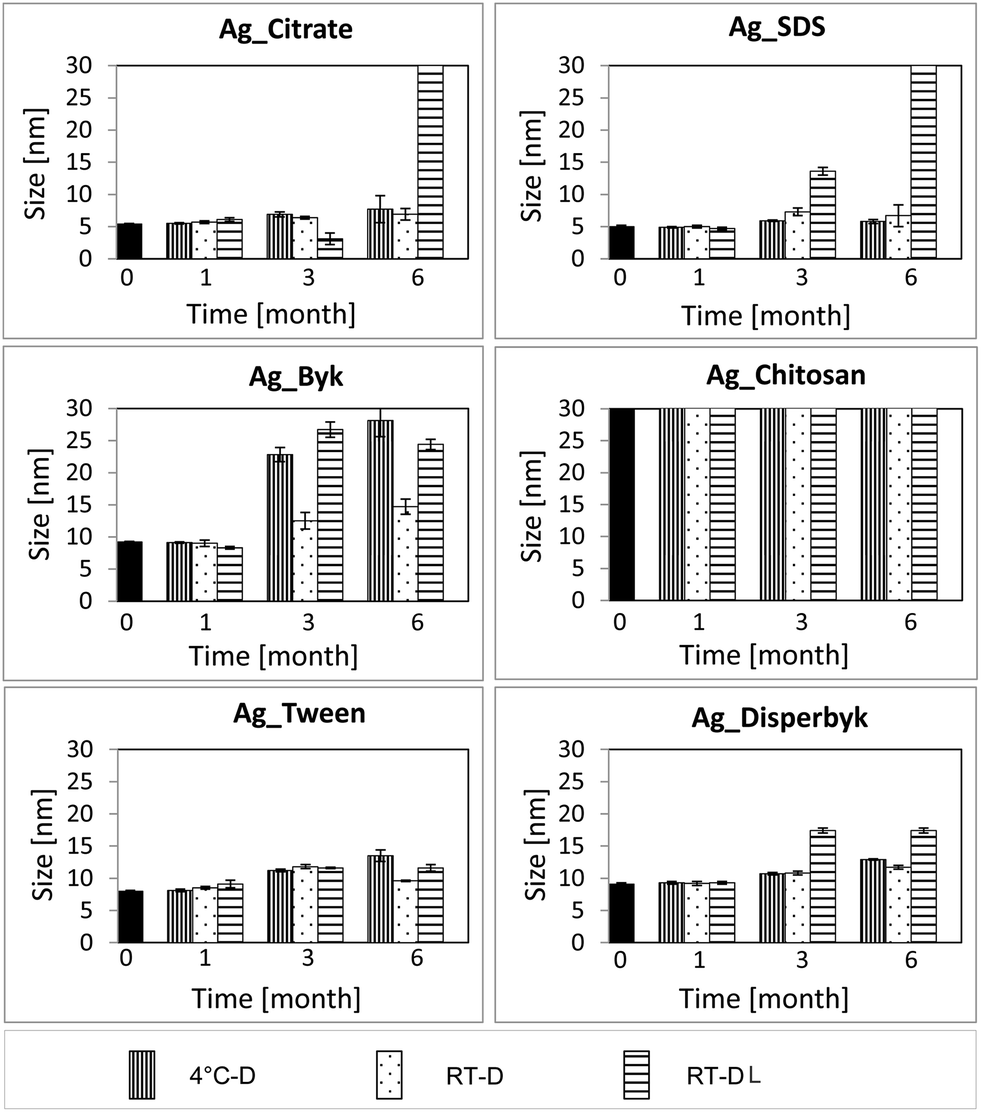 | ||
| Fig. 2 DLS of AgNPs from different time points and storage conditions. | ||
In the case of the positively charged NPs (Ag_Byk), a size change was also detectable in the solutions stored at 4 °C-D (T3 and T6). The neutral particles (Ag_Tween and Ag_Disperbyk) were the least sensitive in terms of size changes resulting from storage.
The UV-vis measurements also showed the strongest deviations at T6, especially in the case of AgNPs stored at RT-DL (Fig. 3). The Ag_Citrate exhibited a slight increase of the absorption maximum and a decrease of the absorbance at 4 °C-D indicative of a slight increase in NP size, while at RT-D the differences were much bigger, confirming additional agglomeration. At RT-DL the absorption maximum could not be determined, suggesting either complete NP dissolution or very significant agglomeration, the latter being confirmed by the DLS data (Fig. 2). The UV-vis signal of Ag_SDS also changed at RT-D and at 4 °C-D, however, the change at RT-D was not as strong as in the case of Ag_Citrate. The highest increase of the absorption maximum and the highest decrease of the absorbance were again observed at RT-DL.
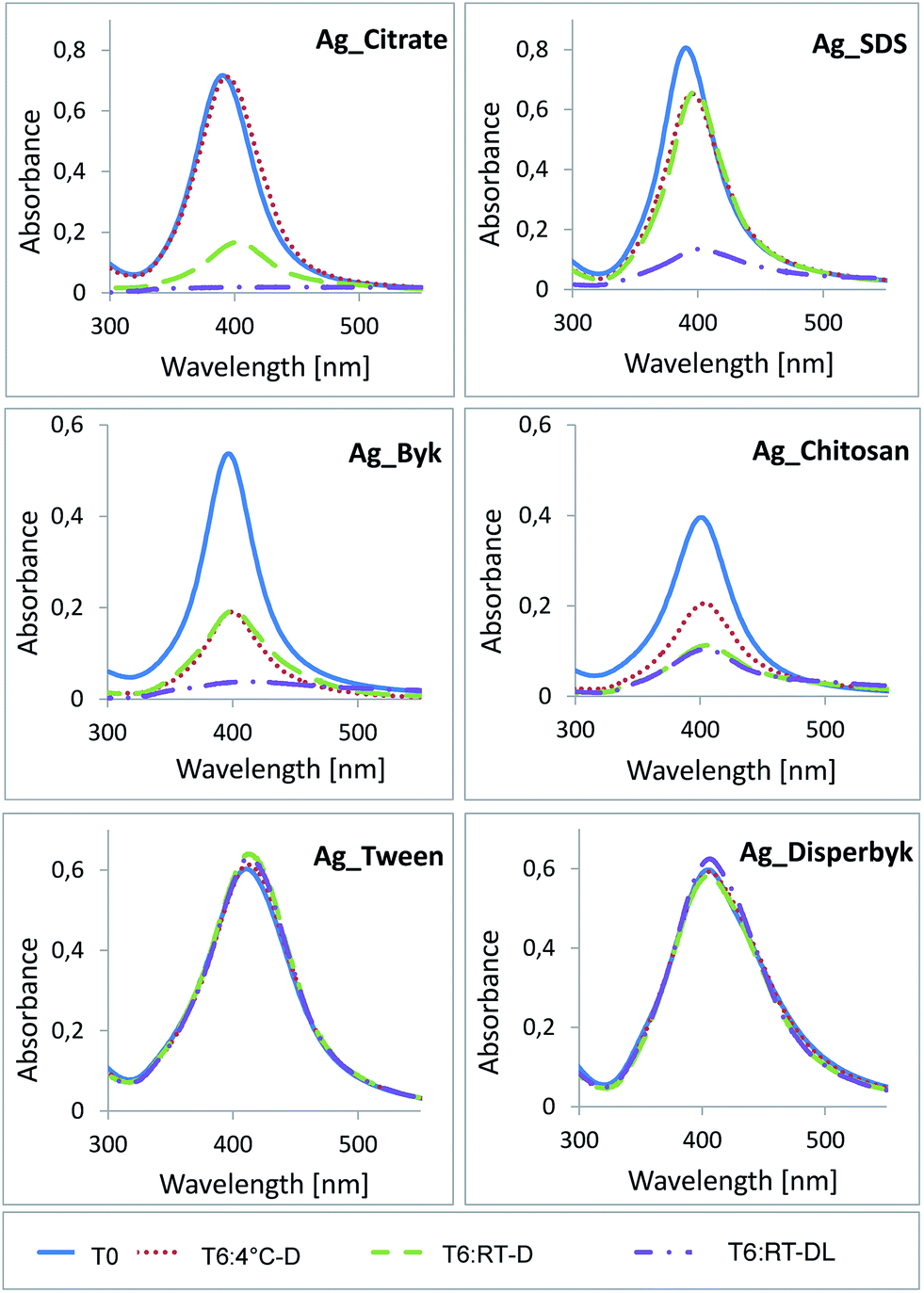 | ||
| Fig. 3 UV-vis of AgNPs from different time points and storage conditions. | ||
In the case of the positively charged NPs, the results were similar to the negatively charged NPs. The Ag_Byk exhibited the most significant deviation at RT-DL. For Ag_Chitosan the most significant shift was observed when kept at RT-D and at RT-DL. The neutral NPs' again showed only some minor shifts.
Changes in surface charge
The zeta potential measurements emerged as an even more sensitive indicator of NP aging, indicating changes in the NPs' surface charges already after 1 month (Fig. 4). In the case of the negatively charged NPs, the zeta potential values dropped significantly after 3 months of storage under all conditions and surprisingly increased after 6 months, especially at RT.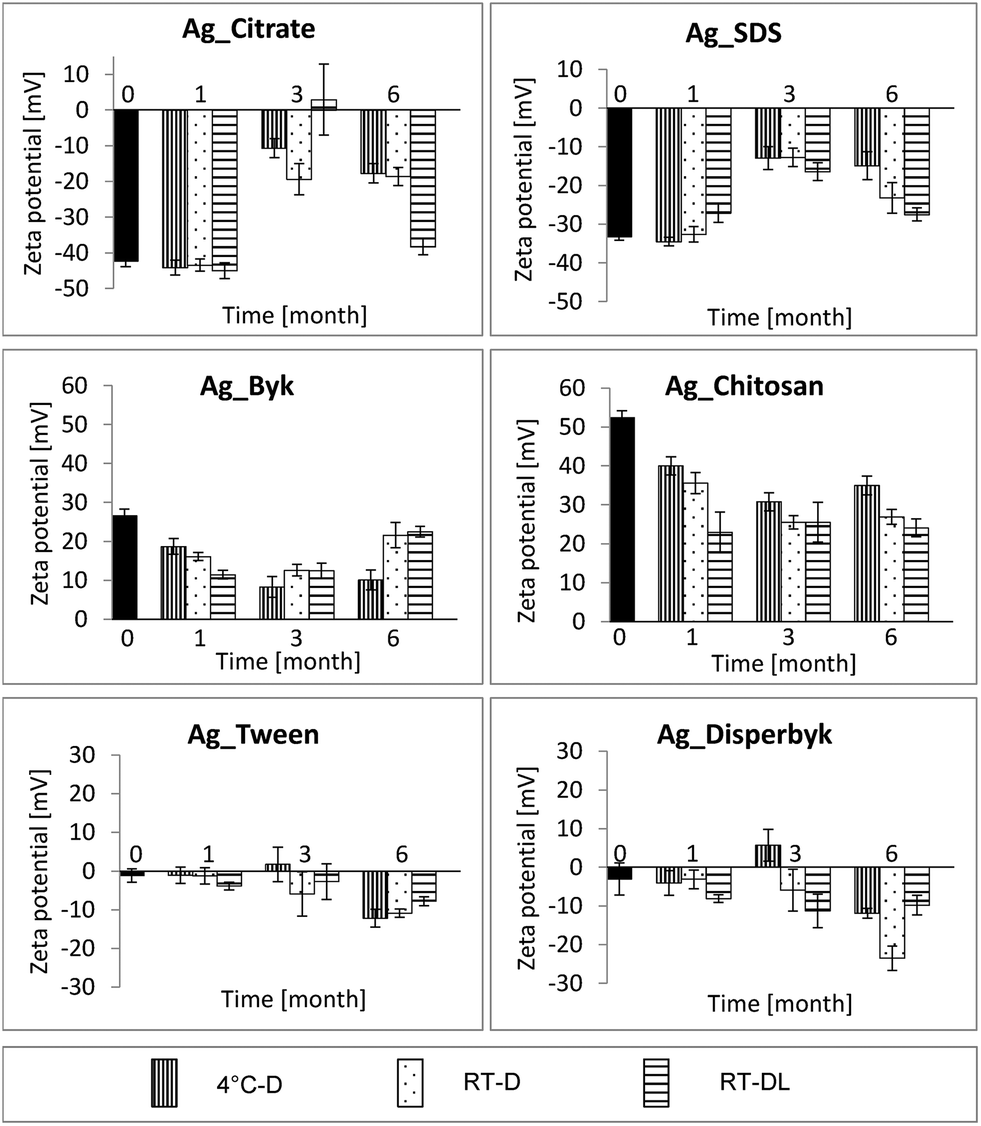 | ||
| Fig. 4 Zeta potential of AgNPs from different time points and storage conditions. | ||
The zeta potential values of the positively charged NPs changed already after 1 month of storage under all conditions, and did not differ significantly in the following months. In the case of the Ag_Byk NPs, the zeta potential value increased slightly after 6 months of storage at RT-D and at RT-DL.
The neutral AgNPs again seemed not to be sensitive to storage conditions and appeared quite stable over the time period studied. There was a change in the zeta potential values mainly after 6 months of storage under all conditions; with both coatings (Ag_Tween and Ag_Disperbyk) the NPs turned more negative, suggesting increased repulsion over time.
Impact of storage time and conditions on Ag NP dissolution
The total amount of AgNPs and Ag+ released was examined by ICP-MS immediately after NP synthesis (T0) and after 6 months of storage (T6:4 °C-D, T6:RT-D, T6:RT-DL). The obtained data are summarized in Fig. 5.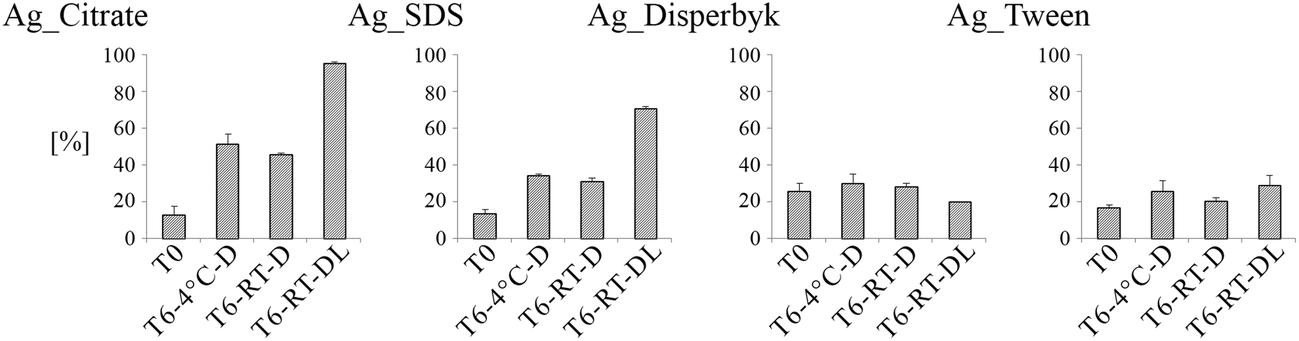 | ||
| Fig. 5 ICP-MS measurements of dissolved Ag from AgNPs at time 0, and after 6 months storage. AgNP samples with different charge and surface composition were stored for 6 months in T = 4 °C without light exposure (T6:4 °C-D); in T = 21 °C without light exposure (T6:RT-D) and in T = 21 °C with light exposure (T6:RT-DL). The amount of released Ag ions was calculated as the difference between the Ag detected at time 0 and 6 months in the extracted fraction. AgNPs with positive charge (chitosan and BYK) were found to interfere with the solid phase of extracted columns and the amount of released silver ions couldn't be estimated for them by this method. | ||
In all cases, a significant amount of Ag+ was detected at T0 (>20% in the case of the Ag_Disperbyk and 10–20% for the Ag_Citrate, Ag_SDS and Ag_Tween). Similar values were detected in another study, where ultracentrifugation was applied to isolate the Ag+ fraction.32
In the case of the negatively charged NPs, longer storage time generally resulted in increased amounts of Ag+, but the storage temperature did not appear to have an impact on the Ag+ release (<5% difference between T6:RT-D and T6:4 °C-D). For the Ag_Citrate stored at RT with exposure to daylight, more than 90% of the Ag was dissolved, while for Ag_SDS under these conditions more than 80% was dissolved. During normal dissolution of AgNPs, an equilibrium state is reached whereby the rates of dissolution and re-precipitation equalize, and consequently further dissolution of AgNPs terminates. In the case when free Ag+ are precipitated (under light exposure), equilibrium cannot be reached and dissolution of AgNPs is not restrained (as can be seen in Fig. 5). As depicted in Fig. 6, the released Ag+ ions precipitated black depositions, most likely Ag2O although this was not confirmed via chemical analysis.33 A 2.5- to 3-fold decrease in the total amount of Ag_SDS or Ag_Citrate in comparison to the initial Ag amount (T0) was observed at T6:RT-DL (Ag_Citrate T0: 11.6 ± 0.3 μg mL−1, T6:RT-DL: 4.4 ± 0.4 μg mL−1; Ag_SDS T0: 12.5 ± 0.1 μg mL−1, T6:RT-DL: 5.1 ± 0.01 μg mL−1), probably due to precipitation of released Ag+ in the form of Ag2O.
 | ||
| Fig. 6 AgNPs stored at: (1) T0, (2) T6:4 °C-D, (3) T6:RT-D, (4) T6:RT-DL. | ||
AgNPs with positively charged coatings were found to bind to the SPE columns, and therefore, the amount of released Ag+ could not be determined by this method. After extraction only <50% of the initial Ag concentration was detected (concentration before extraction: Ag_Chitosan: 10.8 ± 0.4 μg mL−1; Ag_Byk: 11.27 ± 0.27 μg mL−1, concentration after extraction: Ag_Chitosan: 0.04 ± 0.03 μg mL−1; Ag_Byk: 5.2 ± 0.4 μg mL−1).
In the case of the neutral AgNPs, no significant Ag dissolution was observed at any tested storage time or condition, as the determined Ag+ concentrations were similar at T0 and T6 (Fig. 5).
Impact of storage time and conditions on AgNPs cytotoxicity profile
Effects of both negatively charged AgNPs (Fig. 7A) on A549 cell health parameters depended on both storage time and conditions. None of the studied negative AgNPs caused cell death, as it was determined that cell membrane permeabilisation was not statistically different from control samples at either time point. At T6:RT-DL, both Ag_Citrate and Ag_SDS caused very strong acidification and swelling of the lysosomes. The impact of negatively-coated AgNPs on cell number varied a lot between the surface capping agents. Ag_Citrate did not have toxic effects at T6:RT-DL, as it was determined that the number of cells did not decrease (as compared to Ag_Citrate NPs at T0, T6:4 °C and T6:RT-D). Ag_SDS NPs' ability to induce cytotoxicity, measured as a decrease in the cell number, increased with aging, regardless of the storage conditions, which may be a result of Ag dissolution resulting in increasing concentrations of free SDS, which is strongly toxic to cells.40 Both negatively charged AgNPs had a similar impact on the mitochondrial membrane potential following storage, showing an increase at T6:4 °C and especially at T6:RT-D compared to the value at T0.
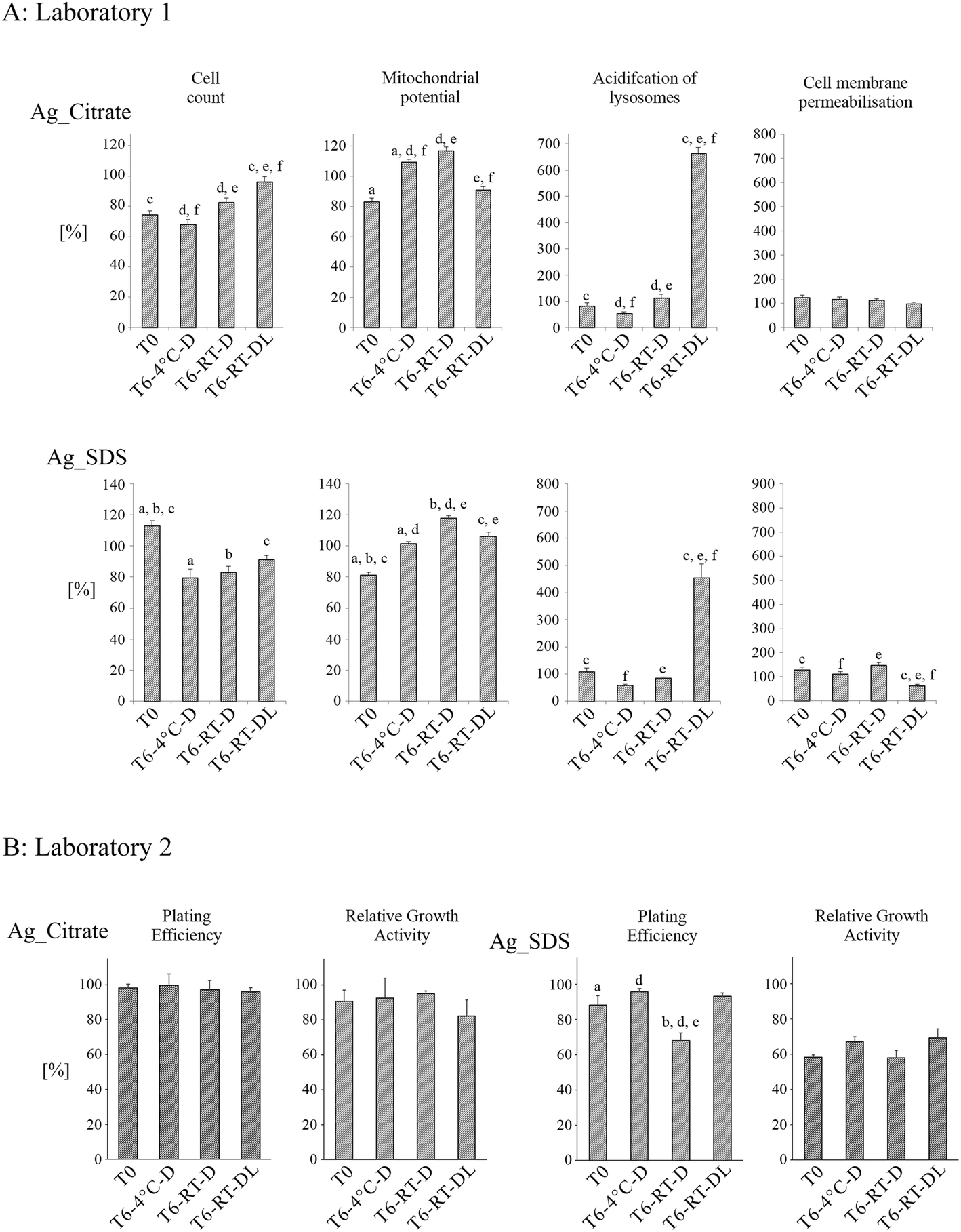 | ||
| Fig. 7 Impact of time and storage conditions on cytotoxicity of AgNPs with negative surface charge (Ag_Citrate, Ag_SDS). AgNPs samples were stored for 6 months in different conditions: T = 4 °C without light exposure (T6:4 °C-D); in T = 21 °C without light exposure (T6:RT-D); in T = 21 °C with light exposure (T6:RT-DL). Cytotoxicity was measured by HCS (Laboratory 1) and by RGA and PE assay (Laboratory 2). Columns represent cytotoxicity relative to 100% of negative control (untreated cells). The data are expressed as mean ± SE of three independent experiments. Letters indicate results between which statistically significant difference has been found (a – T0 and T6:4 °C-D; b – T0 and T6:RT-D; c – T0 and T6:RT-DL; d – T6:4 °C-D and T6:RT-D; e – T6:RT-D and T6:RT-DL; f – T6:4 °C-D and T6:RT-DL). | ||
The positively charged AgNPs (Fig. 8A) caused cell membrane permeabilisation (evidence of cell death) at T0 (Ag_Byk). At T6:RT-DL, the same NPs did not cause cell death but showed the strongest acidification of lysosomes (similar to T6:RT-DL samples of negatively charged AgNPs), suggestive of AgNP dissolution in the lysosomes. The impact on the mitochondrial potential was similar for both Ag_Byk and Ag_Chitosan (a decrease of the mitochondrial potential at T0, T6:4 °C and T6:RT-D compared to the untreated control; and slightly increased or unchanged mitochondrial potential at T6:RT-DL). The cell number was also decreased by treatment with Ag_Byk at T0 and both T6:RT-D and T6:RT-DL (T6:4 °C – no change or increase in cell number). Enhanced toxicity of positively charged NPs is a well-established paradigm, and hence the toxicity at T0 was not surprising, while the fact that the storage at 4 °C seemed to reduce the toxicity over time was unexpected.
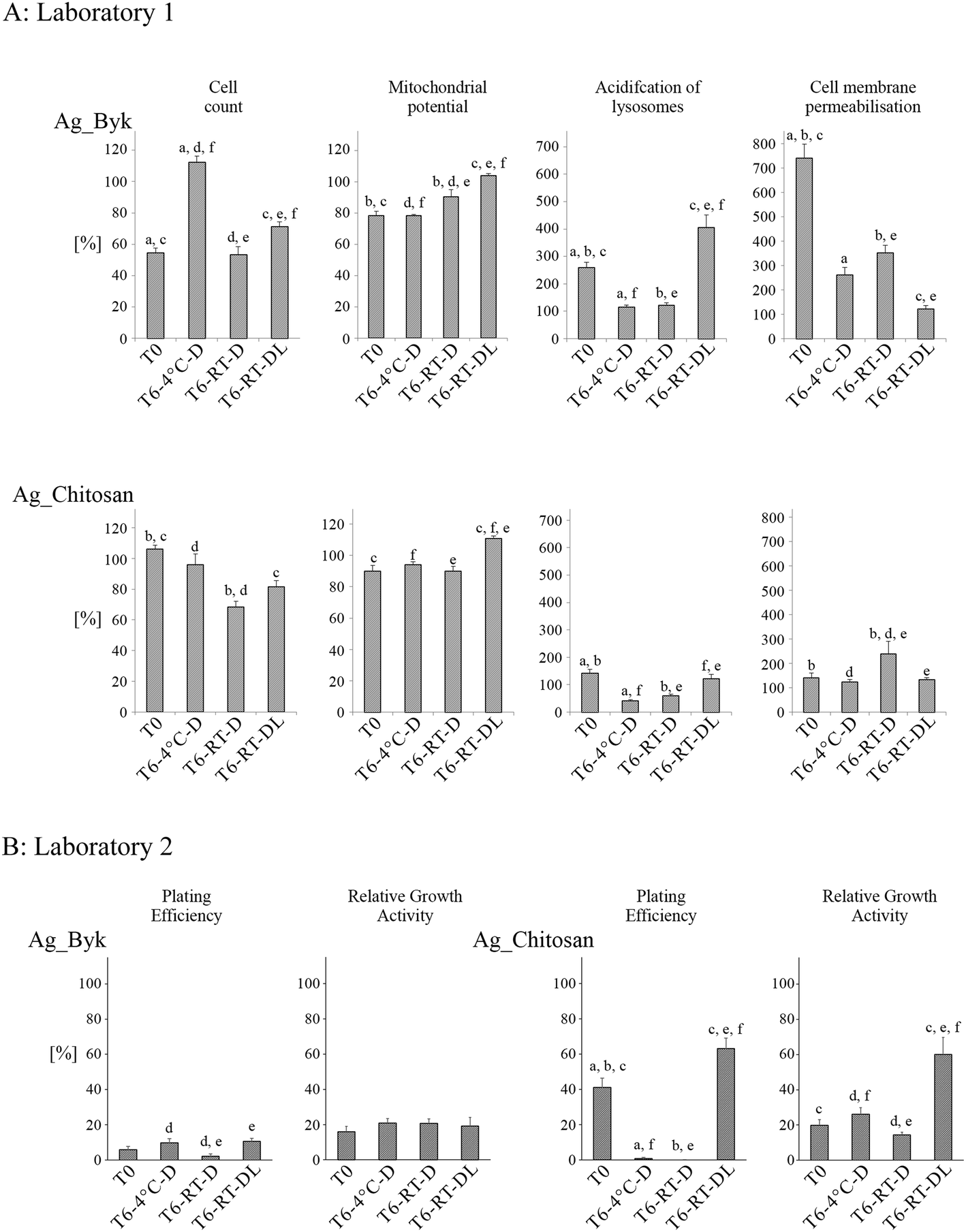 | ||
| Fig. 8 Impact of time and storage conditions on cytotoxicity of AgNPs stabilized by chitosan (Ag_Chitosan) and Byk (Ag_Byk). Ag NPs samples were stored for 6 months under different conditions: T = 4 °C without light exposure (T6:4 °C-D); T = 21 °C without light exposure (T6:RT-D); T = 21 °C with light exposure (T6:RT-DL). Cytotoxicity was measured by HCS (Laboratory 1) and by RGA and PE assay (Laboratory 2). Columns represent cytotoxicity relative to 100% of negative control (untreated cells). The data are expressed as mean ± SE of three independent experiments. Letters indicate results between which statistically significant difference has been found (a – T0 and T6:4 °C-D; b – T0 and T6:RT-D; c – T0 and T6:RT-DL; d – T6:4 °C-D and T6:RT-D; e – T6:RT-D and T6:RT-DL; f – T6:4 °C-D and T6:RT-DL). | ||
The impact of the neutral AgNPs on toxicity endpoints varied less with the storage time and conditions (Fig. 9A) compared to the negatively charged AgNPs. Like the negatively charged AgNPs, neutral AgNPs did not cause cell membrane permeabilisation (no cell death), and acidification of lysosomes was much lower than in the case of either the negatively or positively charged AgNPs (except Ag_Tween: T6:RT-D). In the majority of the samples, the mitochondrial membrane potential remained unchanged or increased slightly compared to the unexposed control suggesting increased reactive oxygen species (ROS) production. The cell count was most affected by the neutral AgNPs. Generally, the Ag_Disperbyk caused a decrease of cell number, and the effect was strongest after 6 months of storage at RT in darkness. In the case of Ag_Tween, a decrease in cell number was found in all samples stored for 6 months.
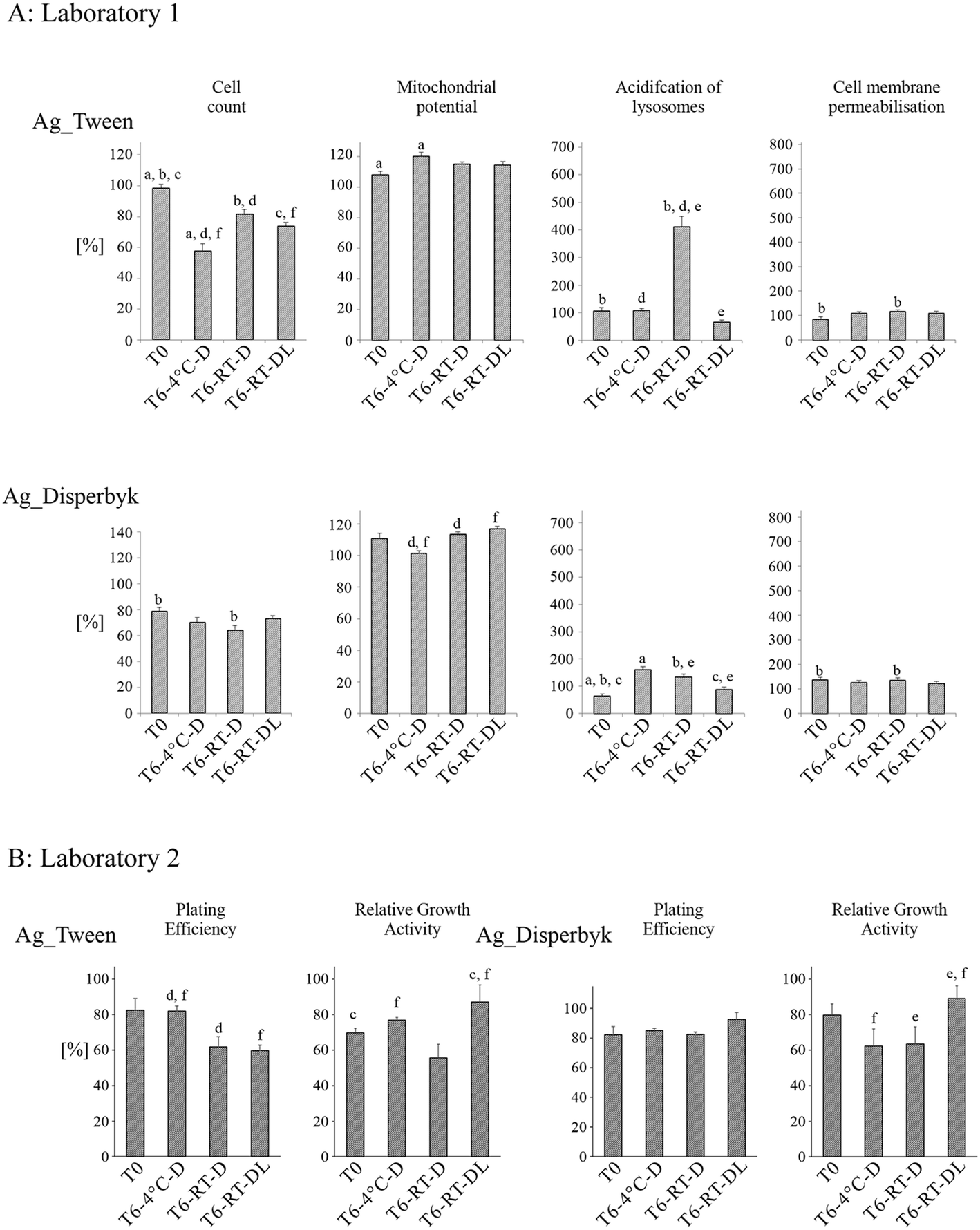 | ||
| Fig. 9 Impact of time and storage conditions on cytotoxicity of AgNPs stabilized by Disperbyk (Ag_Disperbyk) and Tween (Ag_Tween). AgNPs samples were stored for 6 months under the following conditions: T = 4 °C without light exposure (T6:4 °C-D); T = 21 °C without light exposure (T6:RT-D); T = 21 °C with light exposure (T6:RT-DL). Cytotoxicity was measured by HCS (Laboratory 1) and by RGA and PE assay (Laboratory 2). Columns represent cytotoxicity relative to 100% of negative control (untreated cells). The data are expressed as mean ± SE of three independent experiments. Letters indicate results between which statistically significant difference has been found (a – T0 and T6:4 °C-D; b – T0 and T6:RT-D; c – T0 and T6:RT-DL; d – T6:4 °C-D and T6:RT-D; e – T6:RT-D and T6:RT-DL; f – T6:4 °C-D and T6:RT-DL). | ||
The changes in toxicity of the negatively charged AgNPs are summarized in Fig. 7B. In the case of Ag_SDS, the NPs stored at RT-D were found to have significantly higher toxicity than the samples at T0 and the samples stored at 4 °C and at DL. This could be correlated with the higher amount of Ag+ detected in the Ag_SDS sample stored at T6:RT-D as shown in Fig. 5. All tested Ag_Citrate samples were found to be non-toxic in both assays.
The storage time and conditions strongly affected the toxicity of the positively charged AgNPs (Fig. 7B). Toxicity of Ag_Byk, expressed as IC50 values, increased during the 6 months of storage in every case, however, statistically significant differences were not found between the different storage conditions (Table S3†). In the PE assay, only slightly higher toxicity was found in the case of Ag_Byk stored at RT in the dark in comparison to the other samples (T6:4 °C and T6:RT-DL). In the case of Ag_Chitosan, toxicity increased in the samples T6:4 °C (PE assay) and T6:RT-D (PE and RGA assays). At RT-DL, a decrease of toxicity of Ag_Chitosan was observed in both RGA and PE assays at T6. In addition, decreased IC50 values were observed for samples stored in the dark (T6:4 °C and T6:RT-D) and increased IC50 values in the samples exposed to daylight (Table S3†).
In the case of the neutral AgNPs, the cytotoxicity and cell proliferation changed with NP storage time, however, no clear conclusion could be drawn from the results (Fig. 9B). In the RGA assays, Ag_Tween stored at RT-DL were less toxic than the “fresh” AgNPs stock (T0) and the samples stored without light exposure (RT-D). However, in the PE assay, AgNPs stored at RT-D and RT-DL were much more toxic than the same NPs stored at 4 °C. Ag_Disperbyk stored at RT-DL affected cell proliferation and cytotoxicity to a lower extent than the samples T6:4 °C and T6:RT-D. No significant differences were found in the IC50 values for AgNPs coated with Tween and Disperbyk most probably due to their low toxicities (Table S3†).
Discussion
In the last few years, several divergent reports on the toxicity of seemingly the same NPs were published,34–38 as highlighted also by a meta-analysis.26 The inconsistent results could be influenced by the way the materials were stored prior to their toxicological evaluation.39 It is increasingly recognized that NPs' physicochemical properties may change with time and also due to the impact of factors such as temperature and access of light. Thus, the material characterized immediately post-synthesis may not be completely representative of the NPs at the time of the toxicity tests. Understanding the ‘aging’ processes of NPs during storage may thus be crucial for the interpretation of toxicity test results, and may also provide insights into optimal storage conditions for NPs' dispersions in order to optimize their long-term stability and reduce the potential for unnecessary and confounding variations in toxicity. Thus, the main motivation for this study was to investigate changes in NPs' properties occurring over time and to compare the impact of different storage conditions on the observed characteristics, using a panel of AgNPs prepared by an identical method, but with various surface capping agents/charges. Additionally, the impact of the NPs' surface chemistry on their toxicity to in vitro cell cultures was examined, along with assessment of any alteration in the AgNPs' toxicity related to the changes of their physicochemical properties that occurred over time as a result of storing the NPs under different conditions.For the purposes of this study, all AgNPs were synthesized via the same synthesis route and were varied only by the surface-capping agent (Table S1†). Two capping agents per charge were used. The NPs were stored in an excess of the stabilizing solutions, in order to protect them from fast agglomeration. The capping agents were selected for their stabilizating ability and charge, rather than with consideration of their potential toxicity as the primary selection criteria. Some of them, for instance SDS, are well known to be toxic to cells and to increase overall cytotoxicity of NPs.40
Physicochemical characterization – as synthesized
Immediately post-synthesis, full characterization, using a combination of physical and chemical methods, was performed. In general, the full characterization of NPs should include measurements such as particle size distribution, agglomeration state, shape, composition and surface chemistry/charge.41 Thus, the NPs used during this research were extensively characterized using a variety of methods including UV-vis, zeta potential, DLS, TEM, XPS and XRD.As shown by DLS, TEM and XRD measurements, all of the particles are of similar size and shape (Table 1). However, the size measured with the different techniques varied in some cases. In the case of DLS, the NPs' size can differ from the real size because of free surfactant molecules present in the sample, which may change the refractive index of the analyte, or have an impact on NPs' diffusion speed. In the case of TEM, only a small fraction of the material is analysed in comparison to the DLS measurements, so the statistical significance is lower. Additionally, big agglomerates, which are normally detected by DLS, are usually omitted in TEM analysis. It is also difficult to assess aggregation by TEM due to drying artifacts that can result in NP agglomeration during sample preparation. Thus, a large number of TEM images must be assessed in order to have statistical power. The focus here was on comparative analysis of impact of storage conditions rather than absolute size determination. In the case of XRD, the presented size is the average size of single crystals bearing in mind the limitations of this for non-monodisperse samples.
In the case of Ag_Chitosan, it was not possible to analyze the NPs by DLS (the measured size at T0 was more than 1 μm) potentially due to micelle formation of free chitosan.42,43 Since the particle size of Ag_Chitosan analyzed by TEM and XRD was in line with the expected size (TEM: 9.8 ± 2.1 nm, XRD: 7.1 ± 3.1 nm), it can be concluded that the large chitosan molecules (MW: ∼350000 g mol−1), which are normally not detected by TEM and XRD, cause a disturbance during DLS analysis.
Physicochemical monitoring – during storage
TEM, DLS, UV-vis, and zeta potential analysis were applied concomitant with visual inspection to assess the potential changes in the AgNPs' physicochemical properties, which occurred during storage under different conditions. Photographs of the dispersions were taken upon preparation and after 6 months of storage (Fig. 6). These pictures indicated that the strongest transformations occurred in the samples stored at RT and exposed to daylight, which was consistent with previous studies reporting changes of NPs' dispersions kept at ambient atmosphere.44 The significant changes in NPs' properties under this condition could be attributed to the elevated temperature of the dispersions exposed to daylight, which can increase the NP collision rate and subsequently induce faster agglomeration. Additionally, daylight can cause photo-reduction of already dissolved Ag+ that consequently may lead to the production of new NPs increasing the overall sample polydispersity. In the negatively and positively charged AgNPs (T6:RT-DL), there was some evidence of coating degradation, including changes in the colour of the samples and appearance of dark precipitates deposited on the bottom of the storage vials, likely composed of Ag2O.34 The neutral NPs displayed the highest stability, what can be correlated with the size of the stabilizing molecules, the stabilization mechanism (steric versus electrostatic), and the ionic strength and pH of the resulting dispersions. In the case of the positively charged AgNPs, the capping molecules are very big, which may lead to the creation of imperfectly covered NP surfaces. In the uncovered spots, the AgNPs may be easily oxidized and dissolved, inhibiting agglomeration. Additionally, the pH of the positively charged NP dispersions is much lower and the ionic strength higher than in the case of the neutral or negatively stabilized particles, which may also contribute to their faster dissolution/agglomeration. On the contrary, in the case of the negatively charged AgNPs, the capping molecules are very small leading to a very thin surface layer, which could also have contributed to their fast dissolution. The size of the neutral stabilizers seemed to be optimal. Additionally, the neutral NPs were sterically stabilized rendering them less affected by changes in pH or ionic strength, than the electrically stabilized positively and negatively charged NPs.The TEM, DLS and UV-vis measurements allowed similar conclusions as the visual inspection of the stored dispersions. The neutral particles did not exhibit significant changes, even while exposed to daylight at room temperature, while the positively and negatively charged AgNPs agglomerated under this condition. In general, the positively charged NPs were the most sensitive, changing their properties most during storage. UV-vis measurements also showed some changes in the positively charged AgNP solutions kept for 6 months in the dark (at 4 °C and at RT). In general, shifts of the absorption peaks maxima and decreases of absorbance are usually caused by NP dissolution or agglomeration.45,46 However, Yin et al. have shown that a decrease in the absorption intensity and a red shift of the peak maximum may also be caused by NP oxidation.47 Thus, UV-vis measurements are not especially informative as to what is happing but rather indicate that something has changed during storage.
The zeta potential values of the negatively and positively charged AgNPs dropped already after 3 months of storage, also at RT-D and 4 °C-D, and subsequently raised after 6 months of storage, especially at RT-DL. This can be explained by the fact that, in some cases, after 6 months the NPs' agglomerates were big enough to sediment rapidly and the given values came from the particles that had not yet agglomerated. Another reason could be the dissolution process, leading to release of free molecules of capping agents into the solution, and thus the zeta potential value would be a combination of the zeta potential of the NPs and that of the free capping agents.
Dissolution of AgNPs
Given the importance of identifying the free ion fraction for nanotoxicological studies, different methodologies have been applied for the separation of Ag+ from AgNPs: ultracentrifugation, dead-end filtration, flow fluid filtration and dialysis. However, some of these methods are not recommended for use with small NPs.48–51 In our study, SPE was applied in order to separate Ag+ from AgNPs. However, this method was found to be suitable only for neutral and negatively charged AgNPs, as the positively charged AgNPs were trapped in the SPE column. Neutral coating was found to be the most efficient for stabilizing the AgNPs' against dissolution, which is reflected in the stability of the AgNPs' physicochemical characteristics over the 6 months of study, as well as in the cytotoxicity results. Negatively charged AgNPs dissolved faster at RT than at 4 °C, which caused an increase of AgNPs toxicity (in both HCS and RGA for Ag_SDS and HCS for Ag_Citrate).Biological effects of “fresh vs. aged” AgNPs
The observed changes in AgNPs' physicochemical properties resulted in a discernable impact on their toxicity in in vitro cell culture. Two different laboratories performed cytotoxicity tests according to their established assays. Laboratory 1 used HCS, a high-throughput method, to investigate different well-established toxicity endpoints such as cell count, mitochondrial potential, acidification of lysosomes, and cell membrane permeabilisation. It has been previously proven that this method can be successfully applied in nanotoxicology studies and provides several advantages over other approaches such as the multi-factorial analysis and the large numbers of cells/NPs that can be tested in parallel.52,53 In addition to HCS, the toxicity of the AgNPs was measured with two standard cytotoxic tests, PE and RGA, following GLP conditions (Laboratory 2). The RGA and PE assays are efficient and reliable tests for measuring cell proliferation and cell death, and no interference between these methods and tested NPs has ever been observed.54 PE detects both cytotoxic (decrease in number of colonies) and cytostatic effects (smaller size of colonies). The changes in cell number after treatment with AgNPs observed in our study may have been caused by many factors such as the high surface area of NPs, NPs dissolution or the NPs' solvents (dispersants).In the present study, the AgNPs and their aged variants caused different responses in cells. The changes in toxicity of the negatively charged AgNPs depended on both the storage time and conditions. The negatively charged AgNPs stored for 6 months were found to be more toxic than the ‘fresh’ ones. They caused a decrease in the number of cells (Ag_SDS) and, which is most striking, acidification and swelling of lysosomes (T6:RT-DL samples), which presumably precedes lysosomal membrane rupture. This toxicology data, correlated with the higher amount of Ag+ detected in the Ag_SDS and Ag_Citrate samples stored at T6:RT-D (Fig. 5), suggests that the increased concentration of Ag+ exerts the final Ag_SDS (PE) and Ag_Citrate (HCS) toxicity, which is consistent with previous reports.24 However, changes in Ag_SDS toxicity at T6:RT-DL were minor (HCS) or not observed (PE), even though strong dissolution of Ag+ was observed. This was most likely due to conversion of the toxic Ag+ ions into insoluble forms, such as Ag2O (visible as dark deposits in Fig. 6). Some of the harmful effects of Ag_SDS can be related to the presence of a toxic capping agent.55 However, the SDS solvent control (tested at T0) did not show cytotoxic effects at the tested concentration 0.004% w/v (data not shown).31 Thus, Ag_SDS NPs' ability to induce cytotoxicity, measured as a decrease in the cell number (HCS, PE) increased with aging, regardless of the storage conditions, and may be caused by Ag dissolution resulting in increasing concentrations of free SDS.
The positively charged AgNPs were obviously the most toxic ones, and the only ones which (especially Ag_Byk) caused plasma membrane rupture and cell death (HCS, PE, RGA). Enhanced toxicity of positively charged NPs is a well-established paradigm, and thus their toxicity even at T0 was not surprising. However, the fact that storage at 4 °C seemed to reduce the toxicity over time was unexpected. One potential explanation could be the apparent reduction of the effective positive charge over time as evident from the zeta potential data (Fig. 3), resulting in the agglomeration of Ag_Byk (Fig. 2), which has previously been shown to result in decreased cytotoxicity due to reduced uptake.27
The neutral AgNPs were the least toxic regarding changes in cell viability parameters. Still, the observed increase (to various extents) in the acidification of lysosomes after 6 months of storage caused by at least some of the neutral AgNPs stored in the dark, suggested initiation of processes which could potentially lead to cell death. This disturbance of cell viability resulted also in a decreased cell number (caused especially by aged AgNPs). Ag_Disperbyk stored at RT-DL affected cell proliferation and cytotoxicity to a lower extent than the samples T6:4 °C and T6:RT-D, which can be correlated with the enhanced agglomeration of these AgNPs under daylight (Fig. 2).27,36 In addition, in the PE and HCS assays, Ag_Tween stored at RT-D and RT-DL were much more toxic than the same NPs stored at 4 °C. The observed effects can be related to autooxidation of Tween 80® over time, resulting in hydroperoxide formation.56 This degradation process is temperature-dependent and occurs faster at higher temperature, explaining the higher toxicity of samples stored at RT (compared to those stored at 4 °C).
The discrepancies between individual tests can be explained by different cell cultures and exposure conditions such as the number of exposed cells or different CCM and supplements used (Laboratory 1: FCS activated, Laboratory 2: FBS heat inactivated). The differences between the different cytotoxicity assays could also be related to the different endpoints measured.
As shown in this study, several different mechanisms can be involved in AgNP toxicity, such as production of ROS, direct contact of NPs with DNA/chromatin, subcellular dissolution or transporting toxic materials on NPs' surface (the so-called Trojan horse mechanism).35,36 Additionally, some toxic effects can be related to the presence of capping agents or ionic fraction in NPs' samples.46,47 All of these aspects are strictly related to AgNPs' physicochemical properties such as size, number, surface area, charge and chemical composition. We observed that even small changes in NPs' structure could result in different biological responses. Changes in AgNPs' toxicity can be related to solution phase processes such as agglomeration, dissolution, and surface oxidation (insoluble Ag2O on AgNPs' surface) as well as stabilizer degradation. This study highlights the importance of performing a detailed NP dispersion characterization post synthesis and subsequent periodic monitoring of crucial physicochemical parameters over the whole series of replicates, at best before each biological experiment.
Conclusions
The data reported here show clear evidence that AgNPs' ‘aging’ during storage (even under optimal conditions) results in changes in their cytotoxicity, which can be one of the reasons for the contrary results of toxicity reported in the scientific literature for nominally the same materials. The most influential factors of AgNPs' “aging” were found to be higher temperature and exposure to daylight, with the nature of the capping agent and the stabilisation mechanism also contributing. Therefore, a clear and time-resolved understanding of the changes in physicochemical characteristics of any metal/metal oxide NPs occurring under different conditions seems to be crucial for the interpretation of their biological effects.On the basis of the evidence presented here, one important recommendation for nanosafety assessment studies is to periodically monitor the standard NPs' physicochemical parameters such as size/agglomeration, surface charge, and dissolution throughout the duration of the study to ensure that any changes can be accounted for in the data interpretation and analysis. It might also be good practice to note the total time-period during which sets of experiments were performed such that the extent of NPs' aging can be ruled out where all experiments were performed in parallel over a short duration. Researchers should consider also labelling NPs' stock solutions with the date of synthesis & purification (for in house-produced NPs) and the date of arrival and first opening for commercial NPs, to ensure that particles are not be used beyond their manufacturer's guaranteed date, or at least not without verification of their physicochemical integrity.
Acknowledgements
This study was supported by the EU 7th framework program projects: Marie Curie Actions, Network for Initial Training NanoTOES (PITN-GA-2010-264506), QualityNano [INFRA-2010-1.131], Contract no. 214547-2, and NANoREG [NMP.2012.1.3-3], Contract no. 310584 and the Research Council of Norway NorNANoREG project, contract no. 239199/O70. The authors thank Robin Meier (Leonardo da Vinci program) for excellent technical assistance.Notes and references
- P. Gupta, M. Bajpai and S. K. Bajpai, J. Cotton Sci., 2008, 12, 280–286 CAS
.
- K. J. Lee, P. D. Nallathamby, L. M. Browning, C. J. Osgood and X. H. Xu, ACS Nano, 2007, 1, 133–143 CrossRef CAS PubMed
- S. Silver, T. le Phung and G. Silver, J. Ind. Microbiol. Biotechnol., 2006, 33, 627–634 CrossRef CAS PubMed
- T. M. Tolaymat, A. M. el Badawy, A. Genaidy, K. G. Scheckel, T. P. Luxton and M. Suidan, Sci. Total Environ., 2010, 408, 999–1006 CrossRef CAS PubMed
- J. M. Köhler, L. Abahmane, J. Wagner, J. Albert and G. Mayer, Chem. Eng. Sci., 2008, 63, 5048–5055 CrossRef PubMed
- A. Petica, S. Gavriliu, M. Lungu, N. Buruntea and C. Panzaru, Mater. Sci. Eng., B, 2008, 152, 22–27 CrossRef CAS PubMed
- A. D. Russell and W. B. Hugo, Prog. Med. Chem., 1994, 31, 351–370 CrossRef CAS
- L. Yuning, W. Yiliang and S. Beng, J. Am. Chem. Soc., 2005, 127, 3266–3267 CrossRef PubMed
- F. Herzog, M. J. D. Clift, F. Piccapietra, R. Behra, O. Schmid, A. Petri-Fink and B. Rothen-Rutishauser, Part. Fibre Toxicol., 2013, 10, 11 CrossRef CAS PubMed
- H. T. Ratte, Environ. Toxicol. Chem., 1999, 18, 89–108 CrossRef CAS PubMed
- J. H. Sung, J. H. Ji, J. D. Park, J. U. Yoon, D. S. Kim, K. S. Jeon, M. Y. Song, J. Jeong, B. S. Han, J. H. Han, Y. H. Chung, H. K. Chang, J. H. Lee, M. K. Cho, B. J. Kelman and I. J. Yu, Toxicol. Sci., 2009, 108, 452–461 CrossRef CAS PubMed
- A. M. el Badawy, T. P. Luxton, R. G. Silva, K. G. Schekel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS PubMed
- M. Tejamaja, I. Roemer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011–7017 CrossRef PubMed
- J. M. Gorham, A. B. Rohlfing, K. A. Lippa, R. I. MacCuspie, A. Hemmati and R. D. Holbrook, J. Nanopart. Res., 2014, 16, 2339 CrossRef
- S. Elzey and V. H. Grassian, J. Nanopart. Res., 2010, 12, 1945–1958 CrossRef CAS
- J. G. Teeguarden, P. M. Hinderliter, G. Orr, B. D. Thrall and J. G. Pounds, Toxicol. Sci., 2007, 95, 300–312 CrossRef CAS PubMed
- A. R. Jacobson, C. E. Martinez, M. Spagnuolo, M. B. Mcbride and P. Baveye, Environ. Pollut., 2005, 135, 1–9 CrossRef CAS PubMed
- L. Kvitek, A. Panacek, J. Soukupova, M. Kolar, R. Vecerova, R. Prucek, M. Holecova and R. Zboril, J. Phys. Chem. C, 2008, 112, 5825–5834 CAS
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2012, 28, 1095–1104 CrossRef CAS PubMed
- H. S. Shin, H. J. Yang, S. B. Kim and M. S. Lee, J. Colloid Interface Sci., 2004, 274, 89–94 CrossRef CAS PubMed
- M. T. Cisneros, C. V. Ordónez, J. S. Mondragón, A. Campero, O. G. I. Manzano, D. A. M. N. Arrioja, H. P. Mora, A. E. Calderón and I. Sukhoivanov, Microelectron. J., 2009, 40, 618–620 CrossRef PubMed
- A. Shkilnyy, M. Soucé, P. Dubois, F. Warmont, M. L. Saboungi and I. Chourpa, Analyst, 2009, 134, 1868–1872 RSC
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed
- S. Kittler, C. Greulich, J. Diendorf, M. Koller and M. Epple, Chem. Mater., 2010, 22, 4548–4554 CrossRef CAS
- A. K. Suresh, D. Pelletier, W. Wang, J. L. Morrell-Falvey, B. Gu and M. J. Doktycz, Langmuir, 2012, 28, 2727–2735 CrossRef CAS PubMed
- J. A. Varela, M. G. Bexiga, C. Aberg, J. C. Simpson and K. A. Dawson, J. Nanobiotechnol., 2012, 10, 39 CrossRef CAS PubMed
- A. Lankoff, W. J. Sandberg, A. Wegierek-Ciuk, H. Lisowska, M. Refsnes, B. Sartowska, P. E. Schwarze, S. Meczynska-Wielgosz, M. Wojewodzka and M. Kruszewski, Toxicol. Lett., 2012, 208, 197–213 CrossRef CAS PubMed
- H. F. Krug, Angew. Chem., Int. Ed., 2014, 53, 12304–12319 CAS
- V. V. Pintoa, M. J. Ferreira, R. Silva, H. A. Santosc, F. Silva and C. M. Pereirab, Colloids Surf., A, 2010, 364, 19–25 CrossRef PubMed
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC
- A. Huk, E. Izak-Nau, N. el Yamani, H. Uggerud, M. Vadset, B. Zasonska, A. Duschl and M. Dusinska, Part. Fibre Toxicol., 2015, 12, 25 CrossRef PubMed
- S. Agnihotri and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 RSC
- A. de Rooij, ESA Journal, 1989, 13, 363–382 Search PubMed
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2008, 3, 279–290 CrossRef PubMed
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 28 CrossRef PubMed
- A. Huk, E. Izak-Nau, B. Reidy, M. Boyles, A. Duschl, I. Lynch and M. Dušinska, Part. Fibre Toxicol., 2014, 11, 65 CrossRef PubMed
- R. Foldbjerg, P. Olesen, M. Hougaard, D. A. Dang, H. J. Hoffmann and H. Autrup, Toxicol. Lett., 2009, 190, 156–162 CrossRef CAS PubMed
- R. Foldbjerg, D. A. Dang and H. Autrup, Arch. Toxicol., 2011, 85, 743–750 CrossRef CAS PubMed
- M. Kumarasamy, K. Kenesei, Y. Li, K. Demeter, Z. Környei and E. Madarász, Nanoscale, 2015, 7, 4199–4210 RSC
- L. Don, K. L. Joseph, C. M. Witkowski and M. M. Craig, Nanotechnology, 2008, 19, 255702–255707 CrossRef PubMed
- A. B. Stefaniak, V. A. Hackley, G. Roebben, K. Ehara, S. Hankin, M. T. Postek, I. Lynch, W. E. Fu, T. P. Linsinger and A. F. Thünemann, Nanotoxicology, 2013, 7, 1325–1337 CrossRef PubMed
- R. Riva, H. Ragelle, A. des Rieux, N. Duhem, C. Jerome and V. Preat, Adv. Polym. Sci., 2011, 244, 19–44 CrossRef CAS
- N. Zhang, P. R. Wardwell and R. A. Bader, Pharmaceutics, 2013, 27, 329–352 CrossRef PubMed
- H. I. Peng, T. D. Krauss and B. L. Miller, Anal. Chem., 2010, 82, 8664–8670 CrossRef CAS PubMed
- S. Schneider, P. Halbig, H. Grau and U. Nickel, Photochem. Photobiol., 1994, 60, 605–610 CrossRef CAS PubMed
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338–343 CrossRef CAS
- Y. Yin, Z. Y. Li, Z. Zhong, B. Gates, Y. Xia and S. Venkateswaran, J. Mater. Chem., 2002, 12, 522–527 RSC
- E. I. Maurer, M. Sharma, J. J. Schlager and S. B. Hussain, Nanotoxicology, 2014, 8, 718–727 CAS
- M. Hadioui, S. Leclerc and K. J. Wilkinson, Talanta, 2013, 105, 15–19 CrossRef CAS PubMed
- J. C. Trefry, J. L. Monahan, K. M. Weaver, A. J. Meyerhoefer, M. M. Markopolous, Z. S. Arnold, D. P. Wooley and I. E. Pavel, J. Am. Chem. Soc., 2010, 132, 10970–10972 CrossRef CAS PubMed
- G. Dalwadi, H. A. E. Benson and Y. Chen, Pharm. Res., 2005, 22, 2152–2162 CrossRef CAS
- E. Jan, S. J. Byrne, M. Cuddihy, A. M. Davies, Y. Volkov, Y. K. Gun'ko and N. A. Kotov, ACS Nano, 2008, 2, 928–938 CrossRef CAS PubMed
- S. Anguissola, D. Garry, A. Salvati, P. J. O'Brien and K. A. Dawson, PLoS One, 2014, 9, e108025 Search PubMed
- R. Guadagnini, B. H. Kenzaoui, L. Cartwright, G. Pojana, Z. Magdolenova, D. Bilanicova, M. Saunders, L. Juillerat, A. Marcomini, A. Huk, M. Dusinska, L. M. Fjellsbø, F. Marano and S. Boland, Nanotoxicology, 2015, 9(1), 13–24, DOI:10.3109/17435390.2013.829590
- A. S. Inácio, K. A. Mesquita, M. Baptista, J. Ramalho-Santos, W. L. C. Vaz and O. V. Vieira, PLoS One, 2011, 6, e19850 Search PubMed
- B. A. Kerwin, J. Pharm. Sci., 2008, 97, 2924–2935 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10187e |
| ‡ Emilia Izak-Nau, Anna Huk and Bogumiła Reidy contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2015 |