 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow-based pipeline for systematic modulation and analysis of 3D tumor microenvironments†
Cheri Y.
Li‡
a,
David K.
Wood‡
b,
Joanne H.
Huang
c and
Sangeeta N.
Bhatia
*bdefg
aChemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
bHarvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States. E-mail: sbhatia@mit.edu
cBiology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
dBroad Institute, Cambridge, Massachusetts 02142, United States
eDepartment of Medicine, Brigham and Women's Hospital, Boston, Massachusetts 02115, United States
fElectrical Engineering and Computer Science, David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
gHoward Hughes Medical Institute, Chevy Chase, Maryland 20815, United States
First published on 8th March 2013
Abstract
The cancer microenvironment, which incorporates interactions with stromal cells, extracellular matrix (ECM), and other tumor cells in a 3-dimensional (3D) context, has been implicated in every stage of cancer development, including growth of the primary tumor, metastatic spread, and response to treatment. Our understanding of the tumor microenvironment and our ability to develop new therapies would greatly benefit from tools that allow us to systematically probe microenvironmental cues within a 3D context. Here, we leveraged recent advances in microfluidic technology to develop a platform for high-throughput fabrication of tunable cellular microniches (“microtissues”) that allow us to probe tumor cell response to a range of microenvironmental cues, including ECM, soluble factors, and stromal cells, all in 3D. We further combine this tunable microniche platform with rapid, flow-based population level analysis (n > 500), which permits analysis and sorting of microtissue populations both pre- and post-culture by a range of parameters, including proliferation and homotypic or heterotypic cell density. We used this platform to demonstrate differential responses of lung adenocarcinoma cells to a selection of ECM molecules and soluble factors. The cells exhibited enhanced or reduced proliferation when encapsulated in fibronectin- or collagen-1-containing microtissues, respectively, and they showed reduced proliferation in the presence of TGF-β, an effect that we did not observe in monolayer culture. We also measured tumor cell response to a panel of drug targets and found, in contrast to monolayer culture, specific sensitivity of tumor cells to TGFβR2 inhibitors, implying that TGF-β has an anti-proliferative affect that is unique to the 3D context and that this effect is mediated by TGFβR2. These findings highlight the importance of the microenvironmental context in therapeutic development and that the platform we present here allows the high-throughput study of tumor response to drugs as well as basic tumor biology in well-defined microenvironmental niches.
Introduction
The cellular microenvironment, which includes soluble signals such as growth factors and hormones, as well as insoluble signals such as cell–cell and cell–matrix interactions, regulates key aspects of healthy and diseased tissue functions. This observation is particularly relevant in cancer, where the microenvironment has been shown to play a critical role in tumor development, metastasis, and drug resistance.1–4 For example, drug resistance in tumor cells can be modulated by the addition of stromal cells5 as well as culture in 3D spheroids6–9 or encapsulation in a synthetic or natural extracellular matrix (ECM).10,11 The unique phenotypes demonstrated in 3D cell culture are due to changes in a variety of microenvironmental factors, including altered cell–cell contacts, diffusion of nutrients and signaling mediators,12 and integrin ligation with growth factor pathway crosstalk.12–15 Because cellular behavior is dependent on architectural cues, studying microenvironmental influences on cancer progression in 3D could offer unique opportunities. Animal models inherently include critical microenvironmental cues and three-dimensional tissues, but they lack the throughput required for many applications. In vitro tumor models that allow us to control microenvironmental cues specifically in a 3D context may provide a complementary tool to bridge 2D and in vivo studies, and may more accurately predict in vivo cancer progression and response to therapeutics.Systematic exploration of microenvironmental cues for many applications, such as drug screening, requires high-throughput platforms that incorporate rapid production and analysis of combinatorial 3D tissue constructs. Microscale versions (100–500 μm) of cell-laden gels (“microtissues”) can incorporate a range of co-encapsulated stromal and external diffusible cues. Microtissues have been fabricated by various methods including photolithography,16,17 micromolding,18 and emulsification,19 but the majority of these techniques are limited in throughput or result in extremely polydisperse microtissue populations. A promising method for high-speed production of microtissues is droplet-based cell encapsulation, wherein a cell–prepolymer mixture is emulsified on-chip by a shearing oil stream and polymerized while in droplets.20 This process has been demonstrated for a variety of ECM materials, including polyethylene glycol (PEG),20 alginate,21,22 collagen,23 and agarose,24 is compatible with a range of cell types (>90% encapsulation efficiency), and rapidly produces large numbers of monodisperse microtissues (6000 gels min−1). Although droplet devices facilitate high throughput microtissue fabrication, to date analysis of droplet-derived microtissues has relied on serial imaging. While imaging is information-rich, it is labor-intensive and would become a bottleneck in the context of high-throughput screening, especially with large numbers of microtissues. One solution for increasing analytical throughput is the use of an in-flow sorting and analysis system, similar to flow cytometry, that can analyze and sort microtissues on multiple parameters, such as cell density, size and composition based on time-of-flight, extinction, absorbance, and fluorescence. The capability of such a system to quantify fluorescent reporter expression has been demonstrated using microtissues that represent stages of liver development and disease (n ≥ 102–103, fabricated by photolithography).25 Combining high-speed in-flow analysis with a high-throughput microtissue fabrication would produce an ideal system for combinatorial microenvironmental modulation that could be used in high-throughput biology and screening cancer therapeutics.
In this report, we combine microfluidic cell encapsulation with large-particle flow analysis to present an integrated platform for studying the effects of microenvironmental cues (cellular, ECM, growth factors, drugs) on tumor cell proliferation in various 3D contexts. To specifically interrogate the impact of various microenvironmental inputs, tumor and stromal cells were incorporated into droplets at high densities and cell–ECM interactions were controlled by physically entrapping full-length matrix proteins within the encapsulating hydrogel. Furthermore, we leveraged the native stochasticity generated during microfluidic encapsulation to generate diverse subpopulations of microtissues that contain varied degrees of homotypic and heterotypic interactions, and we isolated those subpopulations using flow sorting to generate highly defined microenvironments. As the primary readout, sorted populations cultured with and without exposure to a panel of soluble factors were re-examined via flow analysis to rapidly record large-scale population data (n > 500 events). Finally, we applied this platform to investigate the influence of TGF-β signaling, which is known to be strongly context-dependent and can be either tumor suppressing or cancer promoting, on tumor cell proliferation. We report the outcome of a proof-of-principle drug candidate screen on KrasLSL−G12D/+;p53flox/flox mouse non-small-cell lung cancer (NSCLC) derived cell lines.26 This screen revealed differing sensitivities of these particular lines to TGF-β signaling in 3D that were not observed in 2D. Our ability to study tumor biology and to develop effective new therapies will require systematic study of tumor cells within a microenvironmental context. The platform that we have developed provides a high-throughput method to study drug response and tumor biology within highly-defined microenvironmental niches.
Materials and methods
Tunable microtissue synthesis
Microfluidic device fabrication and cell encapsulation have been described previously.20 Briefly, cells or a mixture of cells were injected into the device as an isopycnic suspension and mixed on-chip with 2× concentrated photopolymerizable polyethylene glycol prepolymer. For microtissues functionalized with matrix proteins, collagen I (rat tail, BD Biosciences), fibronectin (human, Millipore), or laminin (murine, Sigma) were included in the concentrated prepolymer at 40 μg ml−1. The combined aqueous stream, consisting of 10% (w/v) PEG-DA (20 kDa, Laysan), 0.1% (w/v) Irgacure-2959 (Ciba), 1% (v/v) N-vinyl pyrrolidone (Sigma-Aldrich), up to 20 μg ml−1 ECM proteins, and up to 50 × 106 cells ml−1, was sheared into droplets by fluorocarbon oil at a flow-focusing junction. Downstream, ultraviolet light (Exfo Omnicure, 500 mJ cm−2) was used to crosslink droplets into spherical cell-laden hydrogels. Microtissue size was controlled by adjusting the oil/prepolymer flow rates (typically 800 μl h−1 and 200 μl h−1, respectively) to produce monodisperse microtissues between 50–120 μm that were collected and washed in media before preliminary analysis and sorting.Large-particle flow cytometry
Microtissue reporter and cell fluorescence levels were quantified using a complex object parametric analyzer for handling 500 μm objects (COPAS Select, Union Biometrica) according to the manufacturer's instructions. Samples were first gated by time-of-flight (size) vs. extinction (optical density) to exclude cell debris and aggregates. Gated microtissues were then analyzed for Green (gain: 300) and FarRed (gain: 850, −50% Green compensation) fluorescence and sorted into multiwell plates filled with media. Post-sorting, microtissues were washed in PBS by filtering through 40 μm nylon strainers, resuspended in media, and transferred to low-attachment plates for culture and treatment. COPAS data was re-gated and processed using custom MATLAB code.Cell culture
Murine cell lines 393T5 and 394T4 have been previously described.26 Briefly, tumors were initiated in KrasLSL−G12D/+;p53flox/flox mice with intratracheal lentiviral-Cre vectors. Tumors were then excised from the mice, enzymatically digested, and subsequently plated onto tissue culture treated plastic to generate cell lines. Cell lines were transfected with ZsGreen33 and subsequently cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) with 10% fetal bovine serum (Invitrogen), 10 U ml−1 penicillin (Invitrogen), and 10 mg ml−1 streptomycin (Invitrogen). J2-3T3 fibroblasts were cultured in DMEM with 10% bovine serum (Invitrogen), 10 U ml−1penicillin, and 10 mg ml−1 streptomycin. All cells were cultured in a 5% CO2 humidified incubator at 37 °C. To label fibroblasts prior to encapsulation, cells were detached with 0.25% trypsin-EDTA (Invitrogen) and resuspended in PBS. CellTracker Far Red DDAO-SE (Invitrogen, 1.18 mM in DMSO) was added to the cell suspension (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :625 dilution) and incubated for 45 min at 37 °C. The cell pellet was then centrifuged, washed, and either re-plated (the dye was stable for several days) or used immediately.
:625 dilution) and incubated for 45 min at 37 °C. The cell pellet was then centrifuged, washed, and either re-plated (the dye was stable for several days) or used immediately.
Growth factors and inhibitors
Microtissues were cultured in 10% serum media and treated with growth factors EGF, TGF-β, VEGF, or HGF (R&D Systems) at 50 ng ml−1. Small molecule inhibitors were dosed into the microtissue media to a final 10 μM in 0.2% DMSO: SB525334 (Tocris), SJN2511 (Tocris), LY2157299 (Selleckchem), dorsomorphin dihydrochloride (Tocris), DMH-1 (Tocris), or GW5074 (Tocris).Tumor cell proliferation in 2D
393T5 cells were detached with 0.25% trypsin-EDTA (Invitrogen) and replated at a density of 4000 cells/well into 96-well plates. One day post-seeding and daily thereafter, fresh media and drugs were added and ZsGreen fluorescence was measured using a microplate reader (Molecular Devices).Microtissue staining and visualization
Live microtissues were imaged directly for ZsGreen-labeled tumor cell line fluorescence or CellTracker Far Red-labeled fibroblasts. Alternatively, microtissues were fixed and permeabilized in order to count embedded cell nuclei. To bypass any difficulties preserving ZsGreen protein fluorescence after fixation, microtissues containing ZsGreen-labeled cells were additionally incubated with CellTracker Green CMFDA (Invitrogen) prior to being fixed (4% paraformaldehyde). Microtissues were then permeabilized (0.05% Triton X-100), and stained with Hoechst 33342 (Invitrogen). Images were acquired with a Nikon Ellipse TE200 inverted fluorescence microscope, a CoolSnap-HQ Digital CCD Camera, and MetaMorph Image Analysis Software. NIH software ImageJ was used to uniformly adjust brightness/contrast, pseudocolor, and merge images.Statistical analysis
Data are presented as mean ± SEM, except for microtissue cell counts which are described as mean ± standard deviation. Samples were compared using one-way ANOVA, with p-values of <0.05 considered statistically significant.Results and discussion
Platform design
Many techniques for 3D tumor culture have been developed, including encapsulating cells within bulk hydrogels of specific scaffold materials, to control stiffness and ECM composition. However, these systems do not miniaturize readily for high-throughput studies, especially in situations when cells or reagents are limiting. Further, readouts for larger gels often require imaging,15 which is slow and laborious, or biochemical assays that provide only a global measurement averaged over many local microenvironments. Alternatively, formation of 3D tumor spheroids7 has been useful in elucidating the importance of architecture on tumor phenotype. Unfortunately, these niches do not incorporate the kind of microenvironmental control that is available through tuning the physical and biochemical properties of engineered scaffolds.27To generate homogeneous populations of defined microtissues for evaluating proliferative potential under designated microenvironmental and soluble cues, we established an experimental workflow that can be divided into five phases (Fig. 1). First, fluorescently labeled tumor cells are microfluidically encapsulated with the desired combination of stromal cells or ECM into synthetic 3D microtissues (Fig. 1A). We chose poly(ethylene glycol) diacrylate (PEG-DA, 20 kDa) as the hydrogel material because it provides a biocompatible, non-stimulatory background, and unlike other scaffold materials, such as collagen or agarose, PEG can be chemically decorated with integrin binding peptides,28 proteins,29 and other ligands.30
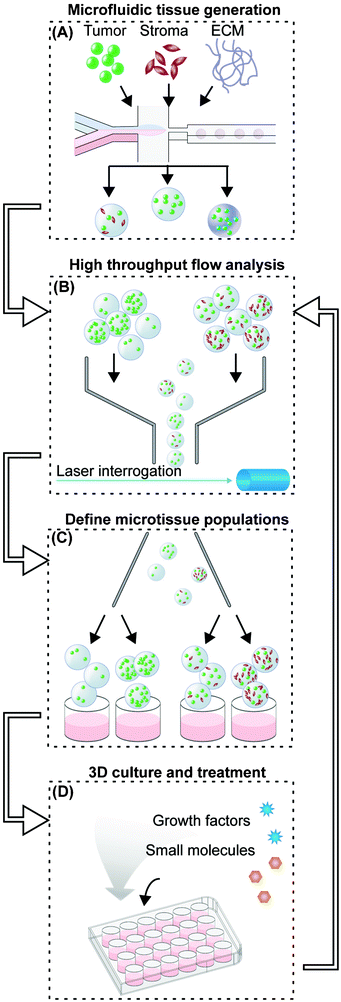 | ||
| Fig. 1 3D tumor microenvironment screening platform. (A) Microfluidic droplet-based encapsulation of tumor cells into microtissues that can be tuned with co-encapsulated stromal cells or entrapped ECM molecules. (B) The microtissues produced are rapidly interrogated in multiple fluorescent channels using large-particle flow analysis. (C) Cytometry-like flow sorting separates and defines microtissues with controlled levels of homotypic and heterotypic interactions. (D) Cellular microenvironment within microtissues is further modulated by soluble factors such as cytokines or small molecule drugs. The extent of cell proliferation within individual microtissues is then detected by flow analysis (B) to collect population-level data on responses to microenvironmental conditions. | ||
In the second phase, a large-particle flow analyzer is used to initially characterize freshly generated microtissues in multiple channels of embedded-cell fluorescence (Fig. 1B). Defined populations of microtissues are selected and sorted by tumor and/or stromal cell density (Fig. 1C). These steps are required because microfluidic cell encapsulation is an inherently stochastic process: for small numbers of cells, a wide range of cell numbers will be encapsulated in each microtissue. In the best-case scenario, theory suggests that the distribution of cells within microtissues will be determined by Poisson statistics.20 However, due to issues of cell settling and aggregation at high cell densities, the cell distribution will often be much more variable in practice. Systems have been optimized to encapsulate single cells,31,32 but controllably encapsulating 10–100 cells, which are closer to the cell density used in spheroid culture,9 is more challenging. While working in this cell density regime, unavoidable variations in microtissue density and composition of different cell types can reduce the statistical power of the analysis. For example, if a microtissue population (n = 500) immediately post-encapsulation has a standard deviation that is 3× the mean fluorescence (σ/μ = 3), one could measure a 40% difference in proliferation with 80% statistical power. Since the population spread usually increases over the course of the experiment due to biological variation, this power would decrease even further for later time points. By contrast, using a pre-sort, initial spreads are constrained to approximately σ/μ = 0.2, with final standard deviations between σ/μ = 0.5 to 1. With these sorted populations, even changes as small as 13% could be detected with 80% statistical power. Further, we take advantage of the initial heterogeneity of the population to produce multiple “bins” of encapsulated cell numbers from a single encapsulation step.
In the next phase, sorted microtissues are collected in tissue-culture wells for culture over 2–6 days, during which time they can be treated with soluble growth factors or drugs (Fig. 1D). During this time, cells proliferate within the microtissues and can be visualized by microscopy. At the desired time point, treated microtissues are collected and re-analyzed by large particle cytometry for changes in overall fluorescence of the embedded cells (Fig. 1B). This method offers higher throughput than methods that require serial imaging as a readout, and unlike traditional bioassays that require release of cells from the microgels, our whole-microtissue flow measurement is non-destructive. After every analysis step using our platform, each microtissue population can be re-collected for additional culture periods and subsequent analysis, allowing us to study the evolution of a single population over time.
Controlling tumor homotypic and heterotypic interactions
Cell–cell interactions, both homotypic and heterotypic, are among the most potent modulators of cellular function. Our platform was designed to generate uniform populations of microtissues of user-defined tumor cell (homotypic) and/or accompanying stromal cell (heterotypic) densities. To demonstrate control over homotypic density, we generated a parent population of microtissues, incorporating a range of numbers of murine non-small cell lung cancer cells (393T5) bearing a constitutive fluorescent reporter protein (ZsGreen). The 393T5 NSCLC cell line was established from a primary tumor that formed distant metastases.26 Because total microtissue fluorescence, as measured using the COPAS, exhibits a linear correlation with cell number (Fig. S1, ESI†), we divided our parent population into multiple subpopulations by enriching each bin for a particular range of encapsulated cells (Fig. 2A). Examination of subpopulations immediately post-sorting reveals three distinctly separated, narrow peaks (Fig. 2A). After three days in culture, cell growth within microtissues yields populations that remained separable, demonstrating the ability to control homotypic density over time (Fig. 2A). During these several days in 3D culture, tumor cells that were originally encapsulated as single cells (Fig. 2B) gave rise to tumor spheroids within the microtissues (Fig. 2C), whereas the same cells typically grow as a monolayer when placed in 2D culture.33 Compared to spheroid models, which require several days to form 3D aggregates, multicellular microtissues in our platform are formed with no time delay. Furthermore, the size and cellular density of spheroids may vary over time due to proliferation and/or contractile forces. The microtissues formed in this study display 3D growth features and allow control of volumetric cell density and interstitial scaffold material.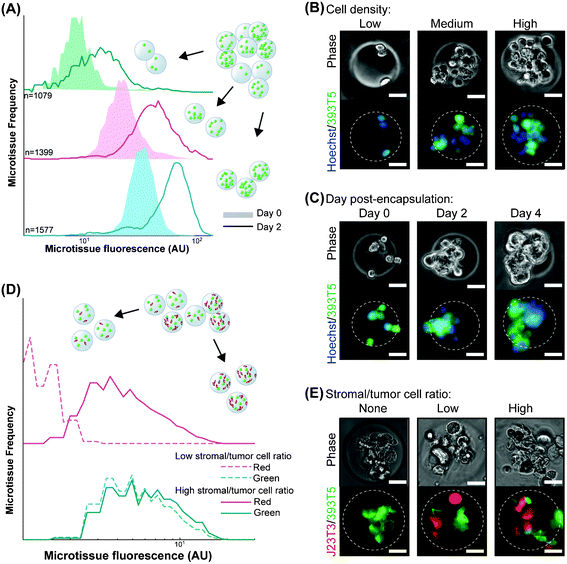 | ||
| Fig. 2 Control over homotypic and heterotypic microtissue composition. (A) Histograms of ZsGreen-labeled 393T5 (lung cancer-derived cell line) microtissue populations, using sorted ZsGreen fluorescence as a measure of homotypic density, before (Day 0) and after (Day 2) proliferation. (B) Phase and epifluorescence images of 393T5 cells embedded within microtissues at various cell densities and stained with CellTracker Green CMFDA. (C) Growth of CellTracker CMFDA stained 393T5 cells within microtissues into spheroids over four days. (D) Microtissues containing 393T5 cells co-encapsulated with CellTracker FarRed stained fibroblasts, sorted by stromal cell density (Red fluorescence) while maintaining desired tumor cell density (ZsGreen fluorescence) to achieve a two-fold change in stromal:tumor cell ratio between the High vs. Low populations. (E) Phase and epifluorescence images of 393T5 cells (ZsGreen) co-encapsulated with J2-3T3 cells at different ratios. All scale bars: 50 μm. | ||
In addition to the influence of homotypic interactions, stromal cells exert a significant effect on tumor growth and the potential for metastasis.1,4,34 In order to study the impact of these cellular interactions, previous studies have varied the stromal cell to parenchymal cell composition within microgels, albeit at lower cell densities, by changing the flow rates of two corresponding cell streams.24 This “pre-encapsulation” control strategy yields the desired stromal:parenchymal cell compositions, at least on average, but the specific ratio in a given microgel varies widely across the population. For example, if two cell types are mixed at a density to give on average 8 cells per gel at a 1:1 ratio, Poisson statistics dictate that only 14% of the resulting gels will actually have equal numbers of the two cells. For an average 1:3 stromal to parenchymal ratio, even fewer gels will contain 1:3 cell numbers, with many gels containing no stromal cells at all.24 To exert finer stoichiometric control of tumor and stroma “post-encapsulation”, we incorporated stromal cells into our microtissue models by mixing and co-encapsulating the 393T5 cells with J2-3T3 murine fibroblasts, and generated a parent population of microtissues from one prepolymer mixture with a range of tumor to stroma ratios. Subsequently, we performed a 2-parameter sort with green and far red fluorescence representing the number of cancer cells and co-encapsulated fibroblasts, respectively (Fig. 2D). We were able to separate the parent population into low (2.5 ± 0.3 cells per gel) and high (5.0 ± 1.7 cells per gel) numbers of fibroblasts, while holding the number of the cancer cells constant (7.0 ± 2.7 cells per gel), thus generating distinct populations with a two-fold range of stromal to cancer ratios, but consistent cancer cell density (Fig. 2E). By defining stromal composition “post-encapsulation” rather than “pre-encapsulation,” we take advantage of the stochasticity of encapsulation to generate multiple populations with different ratios from a single microfluidic process. This allows us to establish populations with a wide dynamic range of absolute cell numbers as well as cellular composition patterns. Further, the tunability of the sorting parameters (Fig. S2, ESI†) allows user-defined tolerances to set the desired spread of cell ratios, which will in general be tighter than those achieved using control over average cell concentrations alone. Therefore, by controlling the bin thresholds, subsequent studies can be performed on populations in which every individual, sorted microtissue contains stromal cells at a particular ratio.
Modulating cell proliferation with microenvironmental factors
At the molecular level, ECM and soluble factors play a large role in modulating cellular function. In cancer, VEGF secretion stimulates angiogenesis, which is a critical component of tumor growth.35 Similarly, matrix remodeling is correlated with a more invasive phenotype.1,36 We were interested in the ability to test how cytokines and ECM modulate metastatic potential in a 3D context, using proliferation as a surrogate for invasive growth. The composition of our PEG-DA hydrogels (10 wt%, 20 kDa) was chosen to form a semi-permeable network (7 nm mesh size37) that allows diffusion of soluble proteins with sizes up to 100 kDa,38 which includes most cytokines. First, we encapsulated 393T5 cells in microtissues, and we sorted them to enrich for a particular homotypic density (17.4 ± 3.4 cells per gel), which we held constant across experiments. Then we cultured the enriched microtissue population for two days in media supplemented with growth factors that have been widely implicated in cancer progression: 50 ng ml−1 of EGF, HGF, VEGF, or TGF-β (Fig. 3A). Exposure to EGF, HGF, and VEGF had no significant effect on proliferation as compared to vehicle control-treated microtissues. The lack of impact of EGF is consistent with the fact that these cells overexpress Kras, which is downstream of the EGF receptor.26,35 Interestingly, treatment with TGF-β led to a significant reduction in proliferation (p < 10−10). TGF-β is known to have a tumor suppressor effect in some early-stage cancers but has also been shown in other cases to promote metastasis, leading to epithelial to mesenchymal transition, especially in later stage cancers.39 While the 393T5 cell line was derived from a primary tumor with proven metastatic potential, our data suggest that the primary tumor still displays an early-stage phenotype that can be suppressed by TGF-β, consistent with observations of other primary lung cancer models.40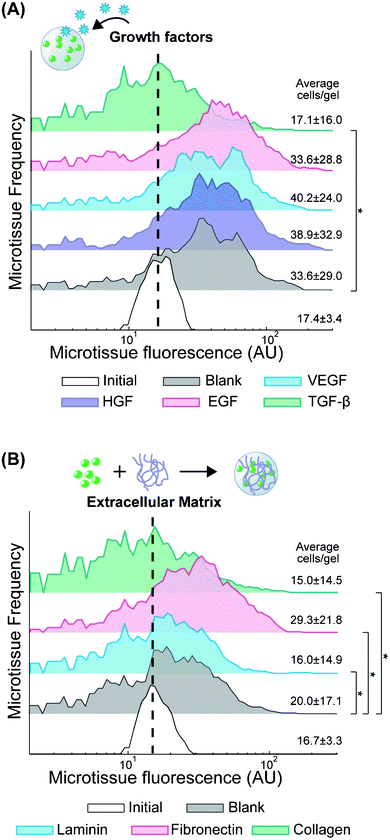 | ||
| Fig. 3 Modulation of tumor cell proliferation by cytokines and ECM. 393T5 growth within microtissues (initial 17.4±3.4 cells per gel) when (A) cultured in media containing 50 μg ml−1 VEGF, HGF, EGF, or TGF-β, or (B) encapsulated in the presence of to 20 μg ml−1 of laminin, fibronectin, or collagen-1 that remain physically entrapped within the hydrogel scaffold. Average number of cells per gel calculated from microtissue fluorescence using linear regression. * indicates p < 0.01. | ||
In addition to examining the impact of soluble factors, we also applied our platform to study the effect of ECM proteins on metastatic potential in 3D. ECM interactions with cell integrins are known to not only trigger direct downstream signaling, but also to modulate the response of cells to other inputs such as drugs and growth factors through pathway crosstalk.41,42 To include ECM in our microtissues, we co-encapsulated 393T5 cells with collagen I (300 kDa), laminin (850 kDa), or fibronectin (440 kDa), adding 20 μg ml−1 of the protein to the pre-polymer mixture so that it is physically incorporated within microtissues during photopolymerization. Due to the size of the hydrogel network, large proteins (>150 kDa) are able to diffuse only very slowly through the gel (Fig. S3, ESI†). Therefore, we expect that the even larger ECM proteins remain effectively entrapped in the microtissues over the timescale of our experiments. Also, at this low concentration, the ECM proteins are unlikely to significantly impact the physical properties of the 100 mg ml−1 PEG-DA hydrogel. Thus, baseline nutrient diffusion and cell growth rates are comparable, allowing a horizontal comparison of ECM molecule signaling effects in 3D using minimal amounts of expensive ECM materials, and without the confounding factor of varying mechanics (e.g. collagen gels vs. fibrin gels) or network properties. ECM-functionalized microtissues enriched for a specific homotypic density were sorted and cultured for 2 days (Fig. 3B). Consistent with their pro-metastatic phenotype, the tumor-derived cells exhibited significantly elevated proliferation in the presence of fibronectin (p < 10−10), which has been shown previously to correlate with metastatic activity.43,44 In contrast, growth was inhibited in the presence of both laminin (p < 0.01) and collagen I (p < 10−4), again demonstrating a tumor cell preference for proliferation in an invasive-supporting matrix over basement membrane proteins. Additionally, collagen I has been reported to induce TGF-β3 expression in some lung cancer cells,45 which could lead to an indirect growth inhibition mediated by this ECM, consistent with our observations in response to TGF-β exposure, described above (Fig. 3A).
Microenvironmental modulation of tumor drug response
Having demonstrated that our sortable microtissue platform can be used to assess the responsiveness of tumor cell populations to soluble as well as embedded matrix proteins, we sought to apply this system to conduct a small-scale pilot drug screen. In contrast to conventional 3D gels, miniaturized tumor microtissues offer an advantage for screening purposes in that reagent costs can be reduced, especially with respect to the amount of drug needed to treat a certain media volume, while the number of replicates is maximized. Combined with its high-throughput readout that will reduce experimental time and effort needed per drug, our platform offers extreme scalability to support even broader screens. As a first proof of concept, we hypothesized that this platform could be used to probe candidate drugs that impact tumor cell proliferation specifically in a 3D architecture, as opposed to any outcomes observed in conventional 2D culture conditions. Given that exogenously supplied TGF-β inhibits 393T5 proliferation (Fig. 3A), and perturbations in TGF-β signaling have been found to be strongly tumor- and context-dependent,39 we selected several small-molecule inhibitors that disrupt aspects of the TGF-β signaling pathway: SB525334 (TGFβR1), SJN 2511 (TGFβR1), and LY2157299 (TGFβR2, TGFβR1). Dorsomorphin (AMPK, ALK2, ALK3, ALK6), DMH-1 (ALK2), and GW5074 (c-raf) were also tested and all treatments were compared to the growth of DMSO vehicle-treated microtissues, or TGF-β as a negative control for 3D growth. For 3D assays, encapsulated 393T5 cells were sorted for a specific population density and cultured for several days in the presence of 10 μM of the inhibitors. Proliferation was assayed based on the change in microtissue fluorescence over time. We compared these results to those found in a 2D assay, where 393T5 cells were seeded on tissue culture microplates and proliferation was tracked by microplate well fluorescence.5Using this assay, we detected statistically significant alterations in microtissue proliferation in response to several of the drug candidates, relative to untreated and DMSO controls (Fig. 4A). The TGFβR1 inhibitor, SB525334, was one of several compounds that exerted similar effects in both 3D and 2D conditions, in that it led to reduced proliferation in each case (Fig. 4B). Dorsomorphin caused cell death in both geometries, and GW5074 elicited little to no anti-proliferative effect (Fig. 4A, B). However, we noted marked differences between 2D and 3D responses to TGF-β and LY2157299. Specifically, while TGF-β inhibited proliferation in 3D as observed previously, the cytokine did not exert any significant effect in 2D. The opposite trend was observed in response to the TGFβR1/TGFβR2 inhibitor, LY2157299, in that it inhibited proliferation in 2D cultures, but did not alter 3D microtissue growth (Fig. 4A, B). We extended our observations by repeating the drug screen using a second cell line isolated from a mouse with the same genetic background (394T4). Consistent results were obtained when the growth responses of drug-treated, sorted microtissues bearing 394T4 cells were compared to 2D cultures (Fig. 4C, D). LY2157299 is a clinically relevant compound undergoing trials for use in a variety of cancer patients,46–48 and has been reported to bind to both receptors, but to TGFβR2 with greater specificity (IC50 2 nM vs. 86 nM for TGFβR1).49 Canonically, TGF-β binds to TGFβR2, which then recruits and phosphorylates TGFβR1. However, it is known that specific TGF-β receptors regulate different activities induced by TGF-β, possibly due to the recruitment of alternative signaling complexes.50 Specifically, several published accounts point to TGFβR2 primarily regulating DNA synthesis, whereas TGFβR1 has been suggested to have a greater impact in mediating matrix synthesis or degradation.51–53 This distribution of functions could be one explanation for why only LY2157299 (inhibiting TGFβR2 for DNA synthesis in addition to TGFβR1) would exhibit the context-dependent but opposing effects on proliferation compared to direct TGF-β treatment, whereas the TGFβR1-only inhibitors (SB525334, SJN2511) did not.
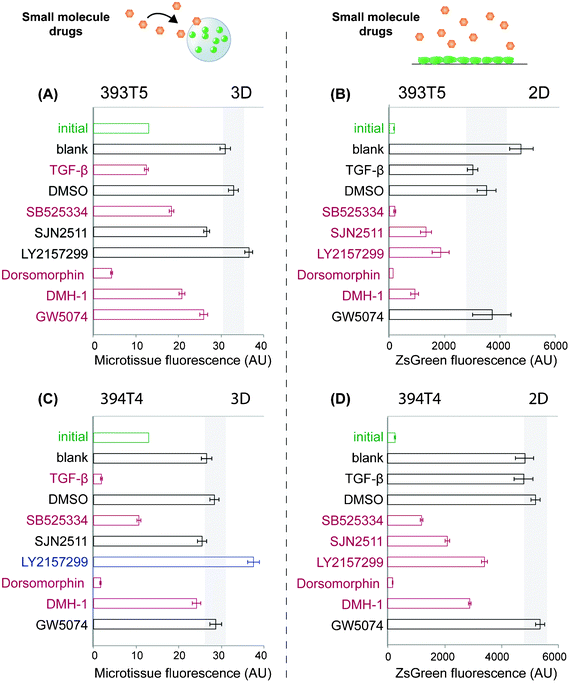 | ||
| Fig. 4 Comparison of 393T5 (A–B) and 394T4 (C–D) lung cancer cell response to drugs when cultured in 3D microtissues (A,C) vs. in 2D monolayers (B,D). Cells were treated in both formats with 10 μM of SB525334 (TGFβR1), SJN 2511 (TGFβR1), LY2157299 (TGFβR2, TGFβR1), Dorsomorphin (AMPK, ALK2, ALK3, ALK6), DMH-1 (ALK2), or GW5074 (c-raf), or 50 ng ml−1 of TGF-β. Microtissue or tissue-culture well fluorescence for each condition are shown after 3 days of culture for 393T5 cells and 5 days of culture for 394T4 cells, which proliferate slower in control conditions, so that the two cell lines undergo the same number of population doublings during each assay. Initial conditions are labeled in green (3D: 13.5 ± 2.5 cells per gel, 2D: 26 × 103 cells cm−2). Gray rectangles indicate the range of p = 0.05 significance by ANOVA with Tukey post-hoc test compared to DMSO controls. Red conditions had significantly reduced cell numbers compared to DMSO controls, whereas blue conditions had significantly increased proliferation rates. | ||
Given the vast, often contradictory, published literature regarding the roles of TGF-β and its receptors, particularly in cancer biology, the impact of drugs may be highly contextual and dependent on tumor models, culture conditions or architectures. This pattern is particularly well-illustrated in our current results and also serves to emphasize the value and importance of evaluating drug candidates in multiple in vitro model systems–perhaps in parallel with established therapeutics in order to calibrate the specific assay readout. In this case, the observation that a TGF-β receptor inhibitor exerts opposing effects on tumor cell proliferation when compared with responses to its ligand is perhaps not unexpected. However, the fact that this same pattern is consistently reversed in our 2D in vitro architecture raises important caveats with respect to the potential responsiveness of tumor cells when this pathway is manipulated in vivo in a clinical setting. Notably, a finding consistent with our result was observed by another group examining a mouse model of metastatic breast cancer.54 In their system, activated TGFβR1 delayed primary tumor growth and accelerated formation of lung metastases, whereas addition of dominant-negative TGFβR2 had the opposite effect. The authors speculate that TGF-β functions as a tumor suppressor in a primary lesion, but promotes metastasis dissemination, which is consistent with our findings that primary-tumor derived lung cancer cells remain responsive to TGF-β stimulation.
Conclusions
We have demonstrated a platform that integrates microfluidic droplet encapsulation to produce microscale tunable microenvironments with a high speed analytical system based on in-flow sorting and analysis of microtissues. This platform leverages tissue engineering materials and methods as well as microfluidic technology, but obviates common problems with 3D tissue engineering constructs, such as laborious fabrication, low-throughput imaging analysis, and low statistical power. The capacity for high-speed analysis enables the detection of hundreds to thousands of individual events in order to assay the impact of microenvironmental conditions on proliferation. Moreover, by sort-based enrichment of defined microtissue populations to limit variability in cell number and/or composition, our platform reduces noise while increasing replicates, which offers the potential to achieve strong statistical significance in biological studies. We used this platform to explore the impact of TGF-β signaling on non-small cell lung cancer proliferation. We demonstrated microenvironmentally-mediated modulation of tumor cell proliferation in this platform and we observed context-dependent signaling via the TGF-β pathway in our model cell lines. Modification of the microtissue scaffold with collagen-1, or treatment of microtissues with TGF-β, diminished cancer cell proliferation uniquely in the 3D setting. Furthermore, a TGFβR1/TGFβR2 inhibitor (LY2157299), but not TGFβR1-only inhibitors, decreased proliferation in 2D yet promoted growth in a 3D context. Based on these results, we predict that the anti-proliferative influence of TGF-β observed in 3D may be mediated by TGFβR2. An interesting extension of these findings would be to conduct a related in vivo preclinical experiment by treating the tumor-prone genetic mouse model,26 which gave rise to our 393T5 and 394T4 lines, with oral LY2157299. Based on our findings, one might predict that the drug might limit or at least delay the appearance of distant metastases, but may not impact the development of primary lung tumors. Future work involving the combined flow-enrichment of subpopulations of microtissues will apply our platform to explore combinations of microenvironmental conditions, such as drug responsiveness in the presence of particular ECM combinations or ratios of heterotypic stromal cell contacts. To support an expanded screen of cues, a microfluidic combinatorial mixer could be incorporated upstream of droplet encapsulation so that ECM and stromal composition could be controlled on-chip. Also, new encapsulation devices including multiple parallel droplet nozzles could augment the microtissue fabrication rate for a full-scale drug screen. Further utility of this platform may be found in extension to other tissues and disease contexts including stem cell or other developmental biology settings, in which the influence of microenvironmental signals has been challenging to study in a methodical, manipulable and screen-compatible fashion.Acknowledgements
We thank Nathan Reticker-Flynn (MIT) for helpful discussions, and Dr Heather Fleming (MIT) for critical readings of the manuscript. We thank Dr Monte Winslow (Stanford) and Dr Tyler Jacks (MIT) for the 393T5 and 394T4 cell lines, and we thank Dr Richard Hynes (MIT) for providing the ZsGreen vector. We thank the MIT Microsystems Technology Laboratories for fabrication support. This work was supported by the Stand Up to Cancer Charitable Initiative from the American Association for Cancer Research. Individual fellowship support was provided by the National Institute of Biomedical Imaging and Bioengineering National Research Service Award fellowship (D. K. W.), the Mazumdar-Shaw International Oncology Fellows Program (D. K. W.), and the National Science Foundation Graduate Research Fellowship Program (C. Y. L., Grant No. 1122374). S. N. B. is an HHMI investigator.References
- P. Friedl and S. Alexander, Cell, 2011, 147, 992–1009 CrossRef CAS.
- J. A. Joyce and J. W. Pollard, Nat. Rev. Cancer, 2009, 9, 239–252 CrossRef CAS.
- B. Psaila and D. Lyden, Nat. Rev. Cancer, 2009, 9, 285–293 CrossRef CAS.
- T. D. Tlsty and L. M. Coussens, Annu. Rev. Pathol.: Mech. Dis., 2006, 1, 119–150 CrossRef CAS.
- R. Straussman, T. Morikawa, K. Shee, M. Barzily-Rokni, Z. R. Qian, J. Du, A. Davis, M. M. Mongare, J. Gould, D. T. Frederick, Z. A. Cooper, P. B. Chapman, D. B. Solit, A. Ribas, R. S. Lo, K. T. Flaherty, S. Ogino, J. A. Wargo and T. R. Golub, Nature, 2012, 487, 500–504 CrossRef CAS.
- P. L. Olive and R. E. Durand, Cancer Metastasis Rev., 1994, 13, 121–138 CrossRef CAS.
- G. Hamilton, Cancer Lett., 1998, 131, 29–34 CrossRef CAS.
- M. Faute, L. Laurent, D. Ploton, M.-F. Poupon, J.-C. Jardillier and H. l. n. Bobichon, Clin. Exp. Metastasis, 2002, 19, 161–167 CrossRef.
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef CAS.
- K. B. Hotary, E. D. Allen, P. C. Brooks, N. S. Datta, M. W. Long and S. J. Weiss, Cell, 2003, 114, 33–45 CrossRef CAS.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. S. Brugge, P. J. Polverini and D. J. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS.
- F. Wang, V. M. Weaver, O. W. Petersen, C. A. Larabell, S. Dedhar, P. Briand, R. Lupu and M. J. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14821–14826 CrossRef CAS.
- E. Rosines, R. V. Sampogna, K. Johkura, D. A. Vaughn, Y. Choi, H. Sakurai, M. M. Shah and S. K. Nigam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20938–20943 CrossRef CAS.
- C. D. Roskelley, P. Y. Desprez and M. J. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12378–12382 CrossRef CAS.
- C. Fischbach, H. J. Kong, S. X. Hsiong, M. B. Evangelista, W. Yuen and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 399–404 CrossRef CAS.
- V. L. Tsang and S. N. Bhatia, Adv. Drug Delivery Rev., 2004, 56, 1635–1647 CrossRef.
- P. Panda, S. Ali, E. Lo, B. G. Chung, T. A. Hatton, A. Khademhosseini and P. S. Doyle, Lab Chip, 2008, 8, 1056–1061 RSC.
- J. Yeh, Y. Ling, J. M. Karp, J. Gantz, A. Chandawarkar, G. Eng, J. Blumling III, R. Langer and A. Khademhosseini, Biomaterials, 2006, 27, 5391–5398 CrossRef CAS.
- C. L. Franco, J. Price and J. L. West, Acta Biomater., 2011, 7, 3267–3276 CrossRef CAS.
- C. Y. Li, D. K. Wood, C. M. Hsu and S. N. Bhatia, Lab Chip, 2011, 11, 2967–2975 RSC.
- V. Trivedi, E. S. Ereifej, A. Doshi, P. Sehgal, P. J. VandeVord and A. S. Basu, Microfluidic encapsulation of cells in alginate capsules for high throughput screening, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2009, 2009, 7037–40 Search PubMed.
- L. Yu, M. C. W. Chen and K. C. Cheung, Lab Chip, 2010, 10, 2424–2432 RSC.
- S. Hong, H.-J. Hsu, R. Kaunas and J. Kameoka, Lab Chip, 2012, 12, 3277–80 RSC.
- E. Tumarkin, L. Tzadu, E. Csaszar, M. Seo, H. Zhang, A. Lee, R. Peerani, K. Purpura, P. W. Zandstra and E. Kumacheva, Integr. Biol., 2011, 3, 653–662 RSC.
- A. A. Chen, G. H. Underhill and S. N. Bhatia, Integr. Biol., 2010, 2, 517–527 RSC.
- M. M. Winslow, T. L. Dayton, R. G. W. Verhaak, C. Kim-Kiselak, E. L. Snyder, D. M. Feldser, D. D. Hubbard, M. J. DuPage, C. A. Whittaker, S. Hoersch, S. Yoon, D. Crowley, R. T. Bronson, D. Y. Chiang, M. Meyerson and T. Jacks, Nature, 2011, 473, 101–104 CrossRef CAS.
- C. K. Choi, M. T. Breckenridge and C. S. Chen, Trends Cell Biol., 2010, 20, 705–714 CrossRef CAS.
- G. H. Underhill, A. A. Chen, D. R. Albrecht and S. N. Bhatia, Biomaterials, 2007, 28, 256–270 CrossRef CAS.
- C. Y. Cheung, S. J. McCartney and K. S. Anseth, Adv. Funct. Mater., 2008, 18, 3119–3126 CrossRef CAS.
- E. Jabbari, Curr. Opin. Biotechnol., 2011, 22, 655–660 CrossRef CAS.
- J. F. Edd, D. Di Carlo, K. J. Humphry, S. Köster, D. Irimia, D. A. Weitz and M. Toner, Lab Chip, 2008, 8, 1262–1264 RSC.
- A. R. Abate, C.-H. Chen, J. J. Agresti and D. A. Weitz, Lab Chip, 2009, 9, 2628–2631 RSC.
- N. E. Reticker-Flynn, D. F. Braga Malta, M. M. Winslow, J. M. Lamar, M. J. Xu, G. H. Underhill, R. O. Hynes, T. E. Jacks and S. N. Bhatia, Nat. Commun., 2012, 3 DOI:10.1038/ncomms2128.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS.
- R. A. Weinberg, The Biology of Cancer, Garland Science, 2007 Search PubMed.
- J. Condeelis and J. W. Pollard, Cell, 2006, 124, 263–266 CrossRef CAS.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287–94 CrossRef CAS.
- A. A. Chen, S. R. Khetani, S. Lee, S. N. Bhatia and J. Van Vliet Krystyn, Biomaterials, 2009, 30, 1113–1120 CrossRef CAS.
- D. Padua and J. Massagué, Cell Res., 2009, 19, 89–102 CrossRef CAS.
- G. Anumanthan, S. K. Halder, H. Osada, T. Takahashi, P. P. Massion, D. P. Carbone and P. K. Datta, Br. J. Cancer, 2005, 93, 1157–1167 CrossRef CAS.
- J. E. Meredith, S. Winitz, J. M. Lewis, S. Hess, X. D. Ren, M. W. Renshaw and M. A. Schwartz, Endocr. Rev., 1996, 17, 207–220 CAS.
- P. M. Comoglio, C. Boccaccio and L. Trusolino, Curr. Opin. Cell Biol., 2003, 15, 565–571 CrossRef CAS.
- S. K. Akiyama, K. Olden and K. M. Yamada, Cancer Metastasis Rev., 1995, 14, 173–189 CrossRef CAS.
- J. Y. Han, H. S. Kim, S. H. Lee, W. S. Park, J. Y. Lee and N. J. Yoo, Lung Cancer, 2003, 41 Search PubMed.
- Y. Shintani, M. Maeda, N. Chaika, K. R. Johnson and M. J. Wheelock, Am. J. Respir. Cell Mol. Biol., 2008, 38, 95–104 CrossRef CAS.
- Eli Lilly and Compan. A Study Combining LY2157299 With Temozolomide-based Radiochemotherapy in Patients With Newly Diagnosed Malignant Glioma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US) 2000- [cited 2012]. Available from: http://http://clinicaltrials.gov/ct2/show/NCT01220271 NLM Identifier: NCT01220271.
- E. Calvo-Aller, J. Baselga, S. Glatt, A. Cleverly, M. Lahn, C. L. Arteaga, M. L. Rothenberg and M. A. Carducci, J. Clin. Oncol., 2008, 26, 14554 Search PubMed.
- Eli Lilly and Company. A Study of LY2157299 in Patients With Hepatocellular Carcinoma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US) 2000- [cited 2012]. Available from: http://clinicaltrials.gov/ct2/show/NCT01246986 NLM Identifier: NCT01246986.
- Selleckchem: LY2157299 Biological Activity [Internet]. Boston (MA): Selleck Chemicals; c2011 [cited 2012]. Available from: http://www.selleckchem.com/products/ly2157299.html.
- B. Bierie and H. L. Moses, Nat. Rev. Cancer, 2006, 6, 506–520 CrossRef CAS.
- R. H. Chen, R. Ebner and R. Derynck, Science, 1993, 260, 1335–1338 CAS.
- R. Derynck and X. H. Feng, Biochim. Biophys. Acta, Rev. Cancer, 1997, 1333, F105–F150 CrossRef CAS.
- M. Centrella, S. Casinghino, J. Kim, T. Pham, V. Rosen, J. Wozney and T. L. McCarthy, Mol. Cell Biol., 1995, 15, 3273–3281 CAS.
- J. M. Yingling, K. L. Blanchard and J. S. Sawyer, Nat. Rev. Drug Discovery, 2004, 3, 1011–1022 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc41300d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |