
An ultrasensitive and incubation-free electrochemical immunosensor using a gold-nanocatalyst label mediating outer-sphere-reaction-philic and inner-sphere-reaction-philic species†
Chiew San
Fang
a,
Kyung Hwan
Oh
a,
Aram
Oh
b,
Kwangyeol
Lee
b,
Seonhwa
Park
a,
Sinyoung
Kim
c,
Jin Kyoon
Park
a and
Haesik
Yang
*a
aDepartment of Chemistry and Chemistry Institute for Functional Materials, Pusan National University, Busan 46241, Korea. E-mail: hyang@pusan.ac.kr; Fax: +82 51 516 7421; Tel: +82 51 510 3681
bDepartment of Chemistry, Korea University, Seoul 02841, Korea
cDepartment of Laboratory Medicine, Yonsei University College of Medicine, Seoul 03722, Korea
First published on 7th April 2016
Abstract
This communication reports a new nanocatalytic scheme based on the facts that the redox reaction between a highly outer-sphere-reaction-philic (OSR-philic) species and a highly inner-sphere-reaction-philic (ISR-philic) species is slow and that an OSR- and ISR-philic Au-nanocatalyst label can mediate the two different types of redox species. This scheme allows highly sensitive and incubation free detection of creatine kinase-MB.
The direct electron transfer between an electrode and a redox species occurs within a few nanometers. This fundamental principle has been widely harnessed to design simple and sensitive electrochemical biosensors.1–3 For example, specific binding of a target (bio)molecule can induce the close approach of a redox label to an electrode along with a conformational change of the redox label-conjugated probe molecule, which in turn allows the direct transfer of electrons, and thereby a high electrochemical signal can be obtained.4,5 However, it is not easy to apply this principle to common sandwich-type biosensors, particularly when the biosensing layer that exists between an electrode and a redox label is not thin enough for direct electron transfer to occur. In the case of redox nanoparticle or catalytic nanoparticle (nanocatalyst) labels, the additional layers (on the labels) that are required to immobilize probe molecules and to minimize the aggregation of the nanoparticle labels make direct electron transfer less feasible.6
Instead of direct electron transfer, one way to obtain fast electron transfer between an electrode and a redox label is to employ an electron mediator that could shuttle electrons between them.7 When a redox enzyme is used as a redox label in a sandwich-type immunosensor, an electron mediator can efficiently shuttle electrons and allow fast enzymatic reactions to occur provided that a redox enzyme label is specifically bound to an immunosensing electrode within a few tens of nanometers.8–10 Contrary to the case of redox enzymes, the mediated electron transfer between a metal nanocatalyst label and an electrode has rarely been investigated. Importantly, it is not easy to simultaneously obtain both a fast redox reaction between an electron mediator and a substrate in the presence of a metal nanocatalyst label and vice versa, especially when the difference between their formal potentials is large.
To obtain a high electrochemical signal-to-background ratio in a biosensor using mediated electron transfer between a nanocatalyst label and an electrode, the following requirements need to be met: (i) the electron mediator should undergo a fast electrochemical reaction at the electrode and a fast catalytic reaction on the nanocatalyst, (ii) the electrochemical reaction of the substrate for the catalytic reaction should be very slow at the electrode, and (iii) the direct reaction (electron transfer) between the electron mediator and the substrate should be very slow, even though the difference in the formal potential between the mediator and the substrate is large.
Redox reactions of inorganic complexes are classified into outer-sphere reactions and inner-sphere reactions,11 and this classification can be extended to redox reactions between an organic electron donor and an acceptor12 and redox reactions at electrodes.13 Furthermore, the authors previously reported that redox species can be classified into species that undergo outer-sphere reactions (OSR-philic species) and species that undergo inner-sphere reactions (ISR-philic species).14,15 Electron mediators such as ferrocene, Fe(CN)63−, the Ru complex, and the Os complex that undergo fast outer-sphere reactions are highly OSR-philic.16 In many cases, the redox reaction between a highly OSR-philic species and a highly ISR-philic species is slow, even though the formal potentials of the two redox species are quite different. For example, the redox reaction between OSR-philic Ru(NH3)63+ and ISR-philic tris(2-carboxyethyl)phosphine is slow.14 Nevertheless, the slow reaction can be accelerated by OSR- and ISR-philic species such as 4-aminophenol: the indirect redox reaction between Ru(NH3)63+ and tris(2-carboxyethyl)phosphine readily occurs via electron mediation of 4-aminophenol.14 Most inner-sphere reactions are slow at indium-tin oxide (ITO) electrodes,17 but many of them are fast at Au electrodes. On the other hand, fast outer-sphere reactions occur well at both ITO and Au electrodes. For these reasons, ITO electrodes can be regarded as OSR-philic species, whereas Au electrodes can be regarded as OSR- and ISR-philic species. If AuNCs are also OSR- and ISR-philic species, they can be used as catalysts that mediate both OSR- and ISR-philic species. Consequently, outer-sphere to inner-sphere redox cycling in the sequence of a highly OSR-philic ITO electrode, a highly OSR-philic electron mediator, OSR- and ISR-philic AuNC, and a highly ISR-philic substrate is feasible (Fig. 1a), which allows the three requirements for obtaining a high signal-to-background ratio to be met.
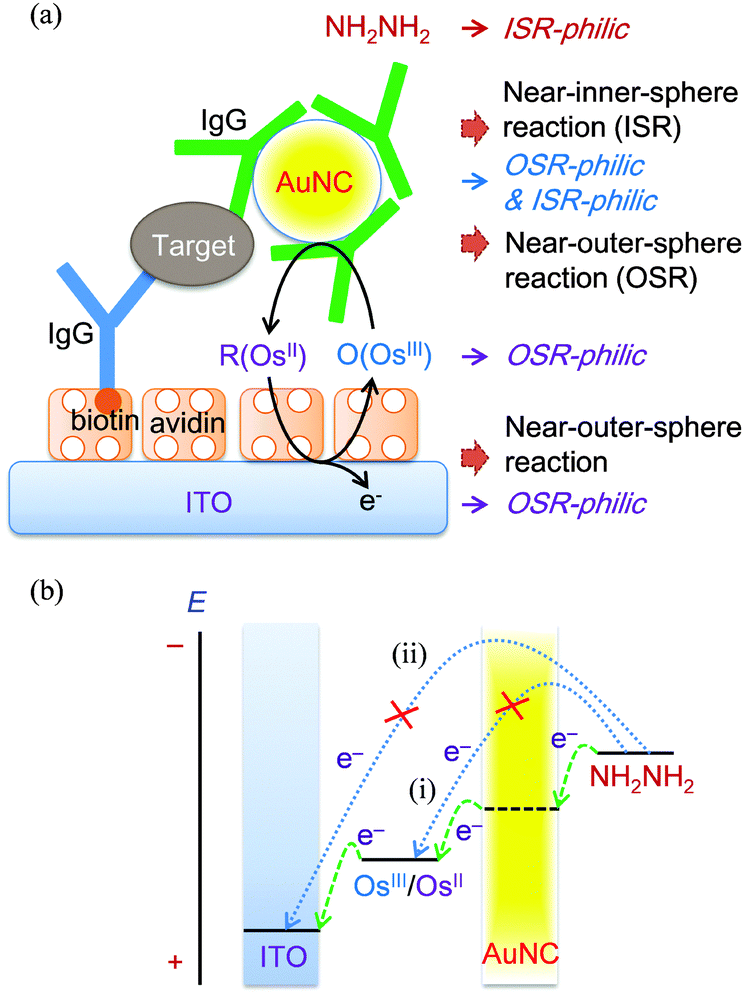 | ||
| Fig. 1 (a) Schematic of an immunosensor using outer-sphere to inner-sphere electrochemical-nanocatalytic (ENc) redox cycling. (b) Schematic of three-step electron transfer related to ENc redox cycling (green lines) and unwanted electron transfer (sky-blue lines). The applied potential, the formal potentials of OsIII(bpy)2Cl2+/OsII(bpy)2Cl2 and hydrazine, and the potential of the Au nanocatalyst (AuNC) (horizontal lines) are positioned in order of potential. | ||
Herein, we report an ultrasensitive, incubation-free electrochemical immunosensor using the outer-sphere to inner-sphere redox cycling that involves an ITO electrode, an electron mediator, an AuNC label, and a strong reductant. Four OSR-philic electron mediators [Ru(NH3)63+, Os(bpy)2Cl2+, ferrocenemethanol (FcMeOH), and Fe(CN)63−] were tested and compared in terms of high signal-to-background ratios. Os(bpy)2Cl2+ was chosen as the optimum electron mediator, and hydrazine, a strong reductant, was used as a substrate for a fast catalytic reaction on AuNC. Electrochemical measurement conditions, including applied potential, concentration of Os(bpy)2Cl2+, and concentration of hydrazine, were optimized. These optimum conditions were applied to a sandwich-type immunosensor for the detection of creatine kinase-MB (CK-MB).18–28
To obtain a high electrochemical signal-to-background ratio in an immunosensor using an AuNC label, the electrochemical signal should be high when the AuNC label is specifically bound to an immunosensing electrode and low when the AuNC label is unbound. For this purpose, it is better to use a low electrocatalytic electrode on which most redox reactions are slow except the redox reaction of an electron mediator that needs to shuttle electrons rapidly between the electrode and the AuNC label. In this regard, an ITO electrode that shows low electrocatalytic activities towards ISR-philic species14,17,29–31 was chosen as a working electrode, and highly OSR-philic metal complexes [Ru(NH3)63+, Os(bpy)2Cl2+, FcMeOH, and Fe(CN)63−] that undergo fast outer-sphere reactions even at ITO electrodes8,9,14,15,32–36 were considered as electron mediators.
It is crucially important to select an appropriate substrate. To obtain a low background level, the direct redox reaction between a substrate and an electron mediator should be slow (arrow (i) of Fig. 1b), and the electrochemical oxidation of a substrate should be slow at the ITO electrode (arrow (ii) of Fig. 1b). For this purpose, it is desirable that the substrate is ISR-philic because the electron mediators and ITO electrodes are OSR-philic.8,9,14,15,17,29–36 To obtain a high signal level, the substrate should rapidly react with the oxidized form of the electron mediator in the presence of AuNC. The substrate should be a strong reductant and have a formal potential much lower than that of the electron mediator. In our previous report, it was shown that the oxidation of hydrazine (a strong reductant) is slow at ITO electrodes and that the redox reaction between hydrazine and a ferrocenium ion is slow.29 Thus, hydrazine was chosen as a substrate.
To obtain a fast catalytic reaction on AuNC, the oxidation of hydrazine (near-inner-sphere reaction) should be fast on AuNC and the reduction of the oxidized form of a highly OSR-philic metal complex (near-outer-sphere reaction) should be fast on AuNC. To meet these requirements, it is desirable that AuNC is both ISR-philic and OSR-philic. Importantly, the electrochemical oxidation of ISR-philic hydrazine (Fig. S1 of the ESI†) and the electrochemical reduction of highly OSR-philic metal complexes are fast at Au electrodes. The results indicate that Au electrodes are ISR-philic and OSR-philic. If AuNCs are also OSR- and ISR-philic species, they can be used as catalysts that mediate both OSR- and ISR-philic species. Both an ITO electrode and an AuNC have high densities of electronic states and can thus give/accept a sufficient number of electrons to/from redox species. Therefore, the potentials of an ITO electrode and an AuNC can be readily tuned. The potential of the AuNC seems to lie between the two formal potentials of Os(bpy)2Cl2+/Os(bpy)2Cl2 and hydrazine, as shown in Fig. 1b.
Fig. 2a shows cyclic voltammograms of the four OSR-philic metal complexes obtained at ITO electrodes in phosphate-buffered saline (PBS). The voltammograms show near-reversible behavior, even at low electrocatalytic ITO electrodes, indicating that electrochemical oxidation and reduction of the metal complexes are fast. The formal potential increases in the sequence of Ru(NH3)63+/Ru(NH3)62+, Os(bpy)2Cl2+/Os(bpy)2Cl2, Fe(CN)63−/Fe(CN)64−, and FcMeOH+/FcMeOH. Fig. 2b represents cyclic voltammograms of the four complexes obtained in the presence of hydrazine at AuNC-modified ITO electrodes. In the case of Os(bpy)2Cl2, highly increased oxidation currents were observed above 0.0 V (curve (ii) of Fig. 2b). The sigmoidal behavior is due to the electrochemical-nanocatalytic (ENc) redox cycling involving the ITO electrode, Os(bpy)2Cl2+/Os(bpy)2Cl2, AuNC, and hydrazine (Fig. 1a). Three-step electron transfer from hydrazine to the ITO electrode occurs (Fig. 1b) during the ENc redox cycling because AuNCs are OSR- and ISR-philic species. On the other hand, the current increase caused by Ru(NH3)62+ was observed at potentials much higher than its formal potential (curve (i) of Fig. 2b). Current increases caused by FcMeOH and Fe(CN)64− (curves (iii) and (iv) of Fig. 2b) were also observed, but the increase occurred at potentials much higher than that of Os(bpy)2Cl2 (curve (ii) of Fig. 2b). To compare the signal-to-background ratios, chronocoulometric data were obtained at 0.16 V in the presence of electron mediators and hydrazine (for measuring background levels) and in the presence of the electron mediator, hydrazine, and adsorbed AuNC (for measuring signal levels) (Fig. S2 of the ESI†). The signal-to-background ratio of Os(bpy)2Cl2+ (624) was the highest among the four metal complexes. Thus, Os(bpy)2Cl2+ was finally selected as the electron mediator.
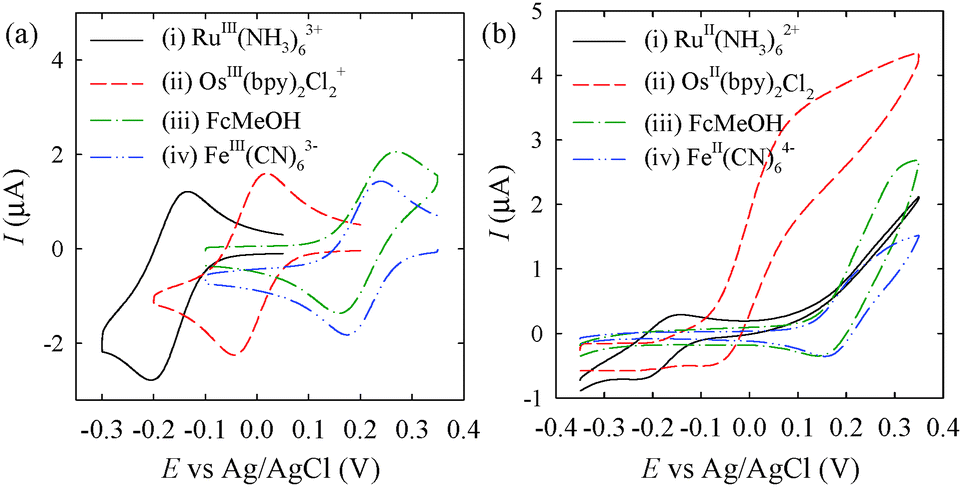 | ||
| Fig. 2 (a and b) Cyclic voltammograms recorded (at a scan rate of 20 mV s−1) using (a) bare ITO electrodes in PBS (pH 7.4) containing 100 μM (i) RuIII(NH3)63+, (ii) OsIII(bpy)2Cl2+, (iii) ferrocenemethanol (FcMeOH), and (iv) FeIII(CN)63− and (b) ITO electrodes modified with adsorbed AuNC, in PBS (pH 7.4) containing 1.0 mM hydrazine and 100 μM (i) RuII(NH3)62+, (ii) OsII(bpy)2Cl2, (iii) FcMeOH, and (iv) FeII(CN)64−. | ||
To investigate the background level in more detail, more cyclic voltammograms and chronocoulograms were obtained and compared (Fig. 3a and c). In the cyclic voltammogram obtained in a solution of hydrazine (curve (i) of Fig. 3a), capacitive currents were mainly observed. The voltammogram obtained in a solution of Os(bpy)2Cl2 (curve (ii) of Fig. 3a) was similar to that obtained in a solution of Os(bpy)2Cl2 and hydrazine (curve (iii) of Fig. 3a). The chronocoulometric signal obtained in a solution of hydrazine (curve (vi) of Fig. 3c) was somewhat higher than that of Os(bpy)2Cl2+ (curve (vii) of Fig. 3c) and lower than that of Os(bpy)2Cl2+ and hydrazine (curve (viii) of Fig. 3c). However, these chronocoulometric signals were much lower than the chronocoulometric signal obtained in the presence of AuNC (Fig. 3d), indicating that the electrochemical oxidation of hydrazine was very slow at the ITO electrode and that the direct redox reaction between Os(bpy)2Cl2+ and hydrazine was also slow. The chronocoulometric signal obtained in a solution of hydrazine in the presence of AuNC (curve (xi) of Fig. 3d) was much higher than that obtained in the absence of AuNC (curve (vi) of Fig. 3c). This indicates that hydrazine is oxidized at AuNC more easily than at the ITO electrode. It is interesting to note that the chronocoulometric signal obtained in a solution of hydrazine and Os(bpy)2Cl2+ in the presence of AuNC (curve (ix) of Fig. 3c) was higher than that obtained in a solution of hydrazine in the presence of AuNC (curve (xi) of Fig. 3c). These results indicate that the ENc redox cycling involving Os(bpy)2Cl2+/Os(bpy)2Cl2 significantly increases the electrochemical signal.
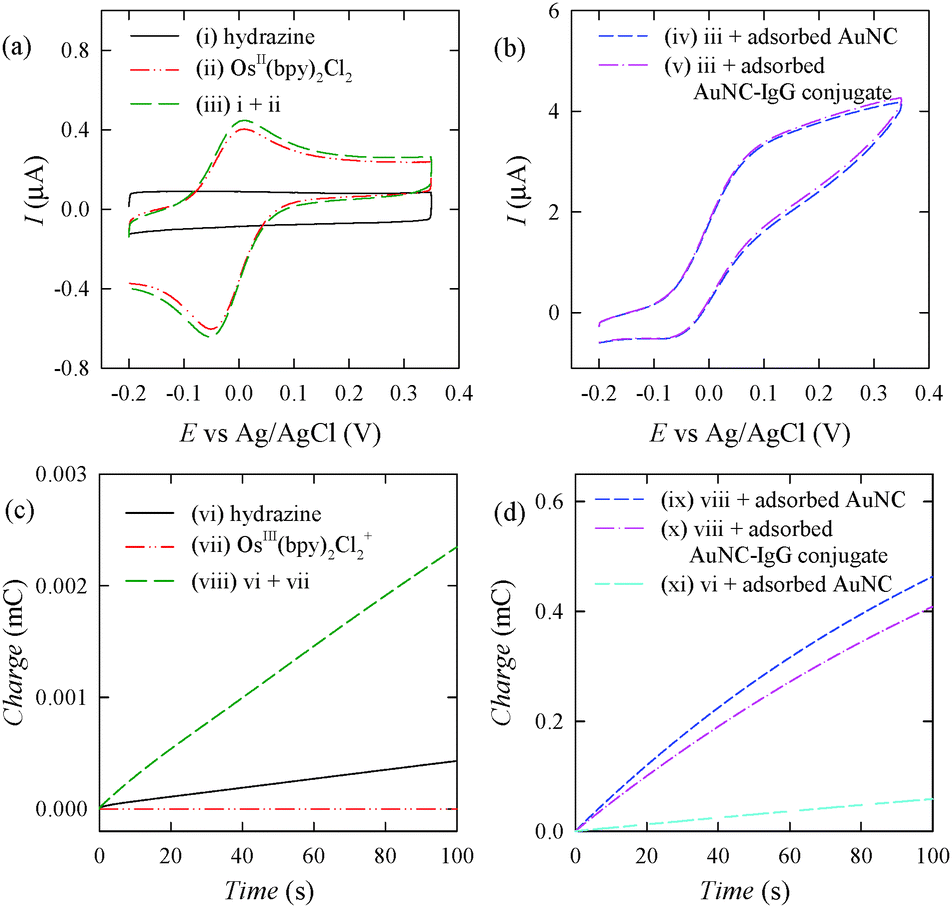 | ||
| Fig. 3 (a and b) Cyclic voltammograms recorded (at a scan rate of 20 mV s−1) using (i, ii and iii) bare ITO electrodes, (iv) an ITO electrode modified with adsorbed AuNC, and (v) an ITO electrode modified with an adsorbed AuNC–antibody conjugate in PBS containing (i) 1.0 mM hydrazine, (ii) 100 μM OsII(bpy)2Cl2, and (iii, iv and v) 1.0 mM hydrazine and 100 μM OsII(bpy)2Cl2. (c and d) Chronocoulograms recorded (at an applied potential of 0.20 V) using (vi, vii and viii) bare ITO electrodes, (ix) an ITO electrode modified with adsorbed AuNC, and (x) an ITO electrode modified with an adsorbed AuNC–antibody conjugate in PBS (pH 7.4) containing (vi) 1.0 mM hydrazine, (vii) 100 μM OsIII(bpy)2Cl2+, and (viii, ix and x) 1.0 mM hydrazine and 100 μM OsIII(bpy)2Cl2+. | ||
The catalytic activity of AuNC may deteriorate significantly after an antibody is adsorbed onto AuNC. To obtain a high electrochemical signal, the catalytic activity of an AuNP-labelled antibody should be similar to that of bare AuNC. The cyclic voltammogram obtained in the presence of adsorbed AuNC (curve (iv) of Fig. 3b) was similar to that obtained in the presence of an adsorbed AuNC-labelled antibody (curve (v) of Fig. 3b). Moreover, there was no significant difference between the chronocoulometric signal obtained in the presence of adsorbed AuNC (curve (ix) of Fig. 3d) and that obtained in the presence of an adsorbed AuNC-labelled antibody (curve (x) of Fig. 3d). These results clearly indicate that most of the catalytic activity of AuNC remained even after the antibody was adsorbed onto AuNC.
The performance of the ENc redox-cycling scheme was evaluated using immunosensors that detect CK-MB and mouse IgG (Fig. 1a). Avidin-modified ITO electrodes were used to immobilize antibodies because the modified electrodes show very low nonspecific binding to many labelled antibodies. The immobilized antibody captures the target and then the AuNC-labelled antibody. The ENc redox cycling involving the ITO electrode, Os(bpy)2Cl2+/Os(bpy)2Cl2, AuNC, and hydrazine occurs upon specific binding of the AuNC-labelled antibody to the captured target. The electrochemical oxidation of Os(bpy)2Cl2 triggers the ENc redox cycling. Consequently, outer-sphere to inner-sphere redox cycling occurs in the sequence of a highly OSR-philic ITO electrode, highly OSR-philic Os(bpy)2Cl2+/Os(bpy)2Cl2, OSR- and ISR-philic AuNC, and highly ISR-philic hydrazine (Fig. 1a). Ultimately, a high electrochemical signal for the nanocatalytic reaction is achieved without an incubation period.
Fig. 4a shows concentration-dependent chronocoulograms obtained at an applied potential of 0.16 V in PBS containing Os(bpy)2Cl2+ and hydrazine after the immunosensing electrodes were treated with PBS containing various concentrations of CK-MB and then with carbonate buffer containing AuNC-labelled anti-CK-MB IgG. The charge data increased with increasing concentration of CK-MB. Fig. 4b shows a calibration plot drawn with the charge data measured at 100 s in the chronocoulograms (Fig. 4a). The calculated detection limit was approximately 20 fg mL−1 for CK-MB, indicating that the immunosensor is highly sensitive and that the nonspecific binding of the AuNC-labelled antibody to the immunosensing electrode is very low. Fig. S3a of the ESI† shows concentration-dependent chronocoulograms obtained after the immunosensing electrodes were treated with PBS containing various concentrations of mouse IgG and then with PBS containing AuNC-labelled antimouse IgG. Fig. S3b of the ESI† shows a calibration plot drawn with the charge data measured at 100 s in the chronocoulograms. The calculated detection limit was approximately 20 fg mL−1 for mouse IgG, reconfirming that the ENc redox cycling allowed ultrasensitive detection. In the case of another cardiac target (myoglobin), the chronocoulometric signal was similar to that of zero CK-MB (Fig. S4 of the ESI†), indicating that the immunosensor is selective.
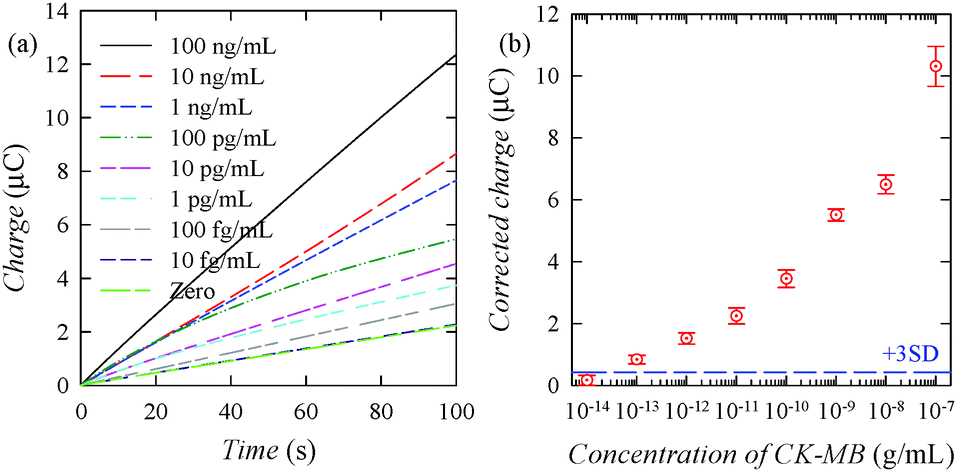 | ||
| Fig. 4 (a) Chronocoulograms obtained (at an applied potential of 0.16 V) using immunosensing electrodes in PBS containing 1.0 μM OsIII(bpy)2Cl2+ and 1.0 mM hydrazine for detecting various concentrations of CK-MB spiked in PBS. (b) Calibration plot: concentration dependence of the charges at 100 s in panel (a). Each experiment was carried out using three different immunosensing electrodes for the assay of the same sample. The data were subtracted by the mean value obtained from seven measurements at zero concentration. The dashed line corresponds to three times the standard deviation (SD) of the charge at zero concentration. | ||
To validate the applicability of the immunosensor using the ENc redox cycling, 15 clinical serum samples were tested using the immunosensor. Each test was carried out using three different immunosensing electrodes for the assay of the same sample, and the charges at 100 s obtained from the chronocoulograms were plotted against the known concentrations of CK-MB (Fig. S5 of the ESI†). The charge increased with increasing concentration of CK-MB with good reproducibility, indicating that the immunosensor is practically appealing.
In conclusion, we have developed a new nanocatalytic scheme using outer-sphere to inner-sphere ENc redox cycling that allows high signal amplification. When the ENc redox cycling was applied to a sandwich-type immunosensor for the detection of CK-MB, the calculated detection limit was approximately 20 fg mL−1, indicating that the immunosensor is highly sensitive. The results for clinical serum samples showed that the immunosensor is practically appealing.
This work was supported by the National Research Foundation of Korea (2015R1A2A2A01002695, 2015M2A8A5021905, and 2012-M3C1A1-048862).
Notes and references
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496 CrossRef CAS PubMed.
- F. Ricci and K. W. Plaxco, Microchim. Acta, 2008, 163, 149 CrossRef CAS.
- K. W. Plaxco and H. T. Soh, Trends Biotechnol., 2011, 29, 1 CrossRef CAS PubMed.
- J. C. Cunning, N. J. Brenes and R. M. Crooks, Anal. Chem., 2014, 86, 6166 CrossRef PubMed.
- J. Hu, T. Wang, J. Kim, C. Shannon and C. J. Easley, J. Am. Chem. Soc., 2012, 134, 7066 CrossRef CAS PubMed.
- J. Das and H. Yang, J. Phys. Chem. C, 2009, 113, 6093 CAS.
- W. Schuhmann, Rev. Mol. Biotechnol., 2002, 82, 425 CrossRef CAS PubMed.
- A. Singh, S. Park and H. Yang, Anal. Chem., 2013, 85, 4863 CrossRef CAS PubMed.
- G. Dutta, S. Kim, S. Park and H. Yang, Anal. Chem., 2014, 86, 4589 CrossRef CAS PubMed.
- G. Dutta, S. Park, A. Singh, J. Seo, S. Kim and H. Yang, Anal. Chem., 2015, 87, 3574 CrossRef CAS PubMed.
- H. Taube, Angew. Chem., Int. Ed., 1984, 23, 329 CrossRef.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641 CrossRef CAS PubMed.
- A. J. Bard, J. Am. Chem. Soc., 2010, 132, 7559 CrossRef CAS PubMed.
- M. R. Akanda, Y.-L. Choe and H. Yang, Anal. Chem., 2012, 84, 1049 CrossRef CAS PubMed.
- H. Yang, Curr. Opin. Chem. Biol., 2012, 16, 422 CrossRef CAS PubMed.
- P. Chen and R. L. McCreery, Anal. Chem., 1996, 68, 3958 CrossRef CAS.
- M. Choi, K. Jo and H. Yang, Bull. Korean Chem. Soc., 2013, 34, 421 CrossRef CAS.
- W. J. Kim, B. K. Kim, A. Kim, C. Huh, C. S. Ah, K. H. Kim, J. Hong, S. H. Park, S. Song, J. Song and G. Y. Sung, Anal. Chem., 2010, 82, 9686 CrossRef CAS PubMed.
- L. Piras and S. Reho, Sens. Actuators, B, 2005, 111–112, 450 CrossRef CAS.
- C. L. Yuan, S. S. Kuan and G. G. Gullbault, Anal. Chem., 1981, 53, 190 CrossRef CAS PubMed.
- A. M. J. Haque, J. Kim, G. Dutta, S. Kim and H. Yang, Chem. Commun., 2015, 51, 14493 RSC.
- J. R. Peela, A. M. Jarari, A. Hai, A. K. Rawal, S. D. Kolla, S. Sreekumar, L. Khurana and N. R. Sidhanathi, Ibnosina J. Med. Biomed. Sci., 2010, 2, 190 Search PubMed.
- A. J. S. Ahammad, Y. H. Choi, K. Kok, J. H. Kim, J. J. Lee and M. Lee, Int. J. Electrochem. Sci., 2011, 6, 1906 CAS.
- E. V. Suprun, A. L. Shilovskaya, A. V. Lisitsa, T. V. Bulko, V. V. Shumyantseva and A. I. Archakov, Electroanalysis, 2011, 23, 1051 CrossRef CAS.
- A. Qureshi, Y. Gurbuz and J. H. Niazi, Sens. Actuators, B, 2012, 171–172, 62 CrossRef CAS.
- M. Pedrero, S. Campuzano and J. M. Pingarron, Electroanalysis, 2014, 26, 1132 CrossRef CAS.
- J. P. Park, M. K. Park and J. W. Yun, Biomarker, 2011, 16, 1 CrossRef CAS PubMed.
- J. Lee, Y. S. Choi, Y. Lee, H. J. Lee, J. N. Lee, S. K. Kim, K. Y. Han, E. C. Cho, J. C. Park and S. S. Lee, Anal. Chem., 2011, 83, 8629 CrossRef CAS PubMed.
- J. Das, K. Jo, J. W. Lee and H. Yang, Anal. Chem., 2007, 79, 2790 CrossRef CAS PubMed.
- M. R. Akanda, M. A. Aziz, K. Jo, V. Tamilvan, M. H. Hyun, S. Kim and H. Yang, Anal. Chem., 2011, 83, 3926 CrossRef CAS PubMed.
- A. N. Asanor, W. W. Wilson and P. B. Oldham, Anal. Chem., 1998, 70, 1156 CrossRef.
- M. R. Akanda, H.-A. Joung, V. Tamilavan, S. Park, S. Kim, M. H. Hyun, M.-G. Kim and H. Yang, Analyst, 2014, 139, 1420 RSC.
- M. R. Akanda, V. Tamilavan, S. Park, K. Jo, M. H. Hyun and H. Yang, Anal. Chem., 2013, 85, 1631 CrossRef CAS PubMed.
- J. Jeong, J. Das, M. Choi, J. Jo, M. A. Aziz and H. Yang, Analyst, 2014, 139, 5814 RSC.
- S. Park and H. Yang, Analyst, 2014, 139, 4051 RSC.
- S. Noh and H. Yang, Electroanalysis, 2014, 26, 2727 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and more supporting data. See DOI: 10.1039/c6cc00353b |
| This journal is © The Royal Society of Chemistry 2016 |