Fragmented polymer nanotubes from sonication-induced scission with a thermo-responsive gating system for anti-cancer drug delivery
Received
27th October 2013
, Accepted 17th December 2013
First published on 18th December 2013
Abstract
Uniform purified long polymer nanotubes with crosslinked poly(glycidyl methacrylate) as the backbone and a pendant poly(N-isopropyl acrylamide) layer attached to the inner surface as a thermo-responsive gating system were synthesized by a two-fold “grafting-from” strategy inside cylindrical alumina nanopores. To adapt such long polymer nanotubes to be efficient nanocarriers for the intracellular delivery of anti-cancer drugs, a sonication-induced scission method was used to ‘cut’ the long nanotubes into short fragments. The discontinuous volume transition property in response to the temperature change for the pendant poly(N-isopropyl acrylamide) layer inside the nanotubes results in a reversible ‘closing/opening’ gating mechanism to control loaded drug release from the nanotubes. Using doxorubicin (DOX) as a model drug, in vitro thermo-responsive drug release behaviour and the related kinetics were studied in detail. The pristine fragmented polymer nanotubes were found to have good biocompatibility in the test with KB-3-1 cancer cells although toxic in their monomeric forms. More importantly, cell toxicity assays for DOX-loaded fragmented polymer nanotubes presented excellent temperature and concentration-dependent cytotoxicity with a low IC50 of 1.4 μmol L−1. These results indicated that such polymer nanotubes with a thermo-responsive gating system have potential as effective anti-cancer drug delivery vehicles.
Introduction
Nonspherical particles have proven to be advantageous for improving the blood circulation time, biodistribution and evading premature clearance by phagocytosis in drug delivery applications.1–5 Development of stimuli-responsive nanotubes, one important type of nonspherical particle, as promising drug carriers has attracted considerable interest because of the enhanced stability of drug molecules over the delivery process, larger drug loading capacity allowing for delivering more than one therapeutic agent, and well-modulated drug release in response to physiological stimulus signals caused by diseases such as temperature or pH changes, redox reactions, enzymes, and so on.6–9 Recently, polymer nanotubes are becoming preferred drug delivery nanovehicles not only because they offer enhanced drug loading capacity and drug stability, but also because they exhibit good potential for surface modification and provide excellent pharmacokinetic control.10–13 To achieve controlled drug release, polymer nanotube-based carriers require reversible stimuli-responsive controlled gating systems, which can prevent drug leakage and deactivation during circulation and allow triggered release of the therapeutics at targeted sites in the presence of physiological stimuli. Inorganic nanoparticles, polymers, and large molecules have been used as blocking gates to attach reversibly to the pore mouths to sterically hinder the loaded drug molecules from releasing.14–16 Different stimuli, such as pH, light, redox potential, temperature, and enzymes, have been applied as ‘triggers’ for uncapping the pores to release the loaded molecules. However, these blocking gates aforementioned were mostly used for mesoporous silica nanoparticle-based drug delivery systems. Hitherto, no gating systems for polymer nanotubes were reported to control their opening/closing to release the guest cargoes.
Successful clinical applications require efficient intracellular delivery of drug-loaded nanotubes through the process of endocytosis during which the nanotubes adsorbed onto the plasma membrane become encapsulated in a vesicle or an endosome that detaches itself from the inside of the cellular membrane and is released into the cytosol.5,11 Both experimental and theoretical investigations have displayed that the cellular uptake of nanostructure carriers was strongly size-dependent.17–20 Nano-sized vesicular systems, ranging from 10 to 1000 nm in size, could be encapsulated by endocytotic pathways through either specific (e.g., receptor-mediated endocytosis) or non-specific interactions (e.g., adsorptive endocytosis) with cell membranes. Therefore, to develop polymer nanotubes into applicable controlled drug delivery systems, thermo-responsive gating uniform nanotubes with suitable size and high purity are highly demanded for efficient cellular uptake via endocytosis.
In this paper, a new type of uniform purified short polymer nanotube with a reversible thermo-responsive controlled gating system was reported which could enhance the drug-loading capacity and sustain drug release in response to the temperature change. The uniform long polymer nanotubes with crosslinked poly(glycidyl methacrylate) (PGMA) as the backbone and the pendant poly(N-isopropyl acrylamide) (PNIPAAm) layer attached to the inner surface as the thermo-responsive gating system (abbreviated as PGMA-PNIPAAm NTs) were synthesized by a two-fold “grafting-from” strategy inside cylindrical alumina nanopores. The purified fragmented short nanotubes were generated by the sonication-induced scission21–24 and dialysis treatment. In vitro drug release studies showed that at ambient temperature with the gating system under closing status, the loaded drugs were preserved within nanotubes without any leakage, while at higher temperature above the lower critical solution temperature (LCST), the gates of the nanotubes were opened, and the loaded drugs were gradually released due to the hydrophobic interaction between the loaded drug and the polymer nanotubes. Moreover, high viability of KB-3-1 cancer cells exposed to pristine PGMA-PNIPAAm NTs was found, which indicated that PGMA-PNIPAAm NTs have good biocompatibility though toxic in their monomeric forms. And in vitro cytotoxicity investigation showed the thermo-responsive and dose-dependent chemotoxicity in fragmented short DOX-loaded polymer nanotubes.
Experimental
Materials and chemicals
Anodic aluminum oxide (AAO) membranes with the nominal diameter of 200 nm and the thickness of 60 μm were purchased from Whatman (Part of GE Healthcare). 3-Aminopropyl trimethoxysilane (APTMS), N,N′-dimethylformamide (DMF), 4,4′-azobis(4-cyanovaleric acid) (ABCA), 1,3-dicyclohexylcarbodiimide (DCC), pyridine, glycidyl methacrylate (GMA), ethylene glycol dimethacrylate (EGDMA), inhibitor remover columns, poly(glycidyl methacrylate) (PGMA, Mn ∼ 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), N-isopropylacrylamide (NIPAAm, 99%), N,N′-methylenebisacrylamide (MBAA) dimethyl sulfoxide (DMSO), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were all purchased from Aldrich, St Louis, MO, USA. Dulbecco's Modified Eagle Medium (DMEM) was purchased from Hyclone Thermo Scientific, Logan, UT, USA. Nanopure water was obtained from Barnstead Easypure RoDi under 18 MΩ cm. Doxorubicin (DOX, Mw = 580) was purchased from MP Biomedicals, LLC, IIIkirch, France.
000), N-isopropylacrylamide (NIPAAm, 99%), N,N′-methylenebisacrylamide (MBAA) dimethyl sulfoxide (DMSO), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were all purchased from Aldrich, St Louis, MO, USA. Dulbecco's Modified Eagle Medium (DMEM) was purchased from Hyclone Thermo Scientific, Logan, UT, USA. Nanopure water was obtained from Barnstead Easypure RoDi under 18 MΩ cm. Doxorubicin (DOX, Mw = 580) was purchased from MP Biomedicals, LLC, IIIkirch, France.
Synthesis of uniform long crosslinked PGMA nanotubes embedded inside AAO nanopores
AAO membranes were washed with acetone by ultrasonication for 10 min and dried under ambient conditions before use. With this membrane as the template, uniform long poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) (poly(GMA-co-EGDMA)) nanotubes embedded inside AAO nanopores were synthesized as described in our previous publication.25 Briefly, the AAO membrane was silanized with APTMS by immersing the template in 1 mM APTMS ethanol solution for 40 min and then the membrane was washed with ethanol and Nanopure water respectively. After blowing with nitrogen gas for several minutes, the AAO membrane was baked in an oven at 120 °C for 30 min, followed by exposure to 40 mL DMF solution containing 0.2 g of photoinitiator ABCA, 2 g of DCC, and 50 mL of pyridine for 8 h at room temperature. The membrane was then cleaned with Nanopure water and acetone respectively, and dried in a vacuum oven at room temperature. Furthermore, GMA and EGDMA were purified by passing through inhibitor remover columns respectively. A precursor material mixture consisting of 70% (v/v) inhibitor-free GMA monomer, 30% (v/v) inhibitor-free EGDMA crosslinker and 1% (w/v) PGMA was homogenized by sonication for 15 min and deaerated by purging with N2 for 10 min. Then the mixture was vacuum-injected into the photoinitiator-immobilized AAO membrane. PGMA nanotubes were generated by the surface-initiated photopolymerization reaction with exposure to UV irradiation for 30 min at 12 mW cm−2 using a DUV Exposure System (ABM Inc., San Jose, CA, USA) fitted with a 500 W HgXe lamp. After the reaction was completed, the nanotubes embedded in the AAO template were dried at 40 °C for 1 h to remove the residual solvent, followed by mechanical polishing of both outer surfaces of the template with 600 grit sand paper to remove the polymer layers. Then the assembly was cleaned with Nanopure water.
Preparation of the thermo-responsive gating system for crosslinked PGMA nanotubes
The reversible thermo-responsive controlled gating system was prepared by attaching the pendant PNIPAAm layer crosslinked with MBAA to the inner surface of crosslinked PGMA nanotubes. Firstly, the monomer NIPAAm was recrystallized from a mixture of 50 mL of toluene and hexane at the volumetric ratio of 4:1. A mixture of 5 wt% of the purified NIPAAm, 2.5 wt% MBAA, and 1 wt% ABCA (dissolved in 0.5 mL THF) in 10 mL Nanopure water was prepared and vacuum-injected into the PGMA-immobilized AAO membrane prepared previously, followed by UV irradiation for 30 min at 12 mW cm−2. Then the ensemble was dried at room temperature for 1 h and polished by 600 grit sand paper, followed by cleaning with Nanopure water. The uniform long crosslinked PGMA nanotubes with the thermo-responsive gating system were released by the dissolution of the AAO template in 3 wt% NaOH solution. After the template is completely dissolved, the free PGMA-PNIPAAm NTs were collected by centrifugation, washed with Nanopure water several times and dispersed in Nanopure water. The amorphous polymer byproducts generated during the formation of nanotubes were removed by dialysis using a cellulose membrane with a cut-off molecular weight of 20000 Daltons (Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA).
Preparation of fragmented short polymer nanotubes with the thermo-responsive gating system
Fragmentation of long crosslinked PGMA nanotubes with the thermo-responsive gating system into short ones was performed by the sonication-induced scission method. An ultrasonic processor (GE × 130, CV18) with a probe diameter of 6 mm working under 130 Watts, 20 kHz was used to sonicate the long polymer nanotubes dispersed in Nanopure water. The sonication time was optimized in the range of 5–60 min. The impurities generated during the ultrasonication process were removed by dialysis using a cellulose membrane with a cut-off molecular weight of 20000 Daltons (Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA).
Anti-cancer drug loading and controlled release
The dried PGMA-PNIPAAm NTs were dispersed in Nanopure water at 5 mg mL−1. DOX was loaded into the PGMA-PNIPAAm nanotubes with the thermo-responsive gating system and tested for the release as follows. 3.68 × 10−3 mol DOX was dissolved in 1 mL DMSO as a stock solution and was diluted to 42.5 μmol L−1 with phosphate buffered saline (PBS; 0.1 M, pH = 7.4). Then the diluted DOX solution was mixed with the pristine PGMA-PNIPAAm NT dispersion solution (5 mg mL−1) at a mass ratio of 1:25 by stirring at 37 °C overnight. Subsequently the DOX-loaded nanotubes were separated by centrifugation and the unloaded DOX concentration was measured using a fluorescence spectrometer (LS-50B, Perkin Elmer) at the excitation wavelength of λex = 480 nm and an emission wavelength of λem = 557 nm. The DOX-loaded nanotubes were then dispersed in 1.8 mL of PBS buffer. The release kinetics were studied with the fluorescence spectrometer on Time-drive mode at both 25 °C and 37 °C maintained by 3006s Isotemp (Thermo Fisher Scientific Inc., Waltham, MA, USA) for 1 h and 10 h, respectively.
Cell toxicity assays
The anticancer activity of the pristine and DOX-loaded PGMA-PNIPAAm nanotubes with the thermo-responsive gating system was evaluated by the MTT method. KB-3-1 cancer cells were cultured in DMEM supplemented with 10% bovine serum at 37 °C in a humidified atmosphere containing 5% CO2 to allow cell attachment. The cells were seeded into four 96-well plates in triplicate at 3000 cells per well. After incubation at 37 °C for 24 h, 20 μL each of the following substances were added: the pristine fragmented short nanotubes (5 mg mL−1) or DOX-loaded fragmented short nanotubes with the loaded DOX concentration ranging from 0.1 to 10 μmol L−1. After 72 h incubation, the MTT solution (4 mg mL−1, 20 μL) was added to each well and the cells were further incubated for 4 h. Finally, the supernatant was removed and 100 μL DMSO was added into each well to dissolve the formazan crystals with vortex mixing. The absorbance of each well was determined using an OPSYS Microplate Reader (DYNEX Technologies, Inc., Chantilly, VA) at 570 nm. The concentrations required to inhibit cellular growth by 50% (IC50) were then calculated from survival curves.
Characterization and analysis
The morphologies of the long and fragmented short PGMA-PNIPAAm nanotubes were imaged by Scanning Electron Microscopy (SEM) (TM-1000, Hitachi High Technologies America, Inc., Pleasanton, CA) at an accelerating voltage of 15 kV and High Resolution Electron Microscopy (HRTEM) (JEOL, Peabody, MA, USA) at an accelerating voltage of 200 kV. A drop of each sample was placed on the SEM stub and dried under ambient conditions. To prevent charging of the specimen with an electron beam, the Polaron SC7620 mini Sputter Coater (Quorum Technologies Ltd, East Sussex UK) was employed to coat the specimen with a 10 nm thin layer of Pd/Au. For HRTEM analysis, samples were dispersed in ethanol and placed on copper grids.
Results and discussion
Uniform long crosslinked polymer nanotubes with the thermo-responsive gating system
The uniform long polymer nanotubes with crosslinked PGMA as the backbone and the pendant PNIPAAm layer attached to the inner surface of the PGMA nanotubes as the thermo-responsive gating system were synthesized from a two-fold “grafting-from” strategy as shown in Fig. 1. The first-fold was the synthesis of uniform long crosslinked PGMA nanotube backbones embedded in cylindrical alumina nanopores using a surface-initiated UV-polymerization pattern transfer method.25,26 This process includes silanization, photoinitiator immobilization, vacuum injection of precursor materials, UV-polymerization, and mechanical polishing. Silanization involves the modification of the AAO template surfaces with APTMS. The Si–O bond formed between the hydroxyl-terminated surfaces of the AAO template and APTMS with the bond energy of 452 kJ mol−1 is very stable even at a high temperature, thus enhancing the stability. Next, the photoinitiator, ABCA, was immobilized onto the surfaces of the activated AAO template via amide bond formation between carboxylic groups of ABCA and amine functional groups of APTMS. Then the precursor material consisting of the GMA monomer, EGDMA cross-linker and pre-synthesized PGMA polymer was vacuum-injected into the photoinitiator-immobilized AAO nanopores. The incorporation of the pre-synthesized polymer into the monomer solution as the precursor material allows for precompensation of the structure shrinkage induced by photopolymerization reactions.25 Finally crosslinked PGMA nanotube backbones embedded in cylindrical alumina nanopores were formed by free radical photo-polymerization under the effect of surface-bound initiators followed by the mechanical polishing to remove the outer polymer layers. The second-fold of the “grafting-from” strategy as shown is photo-polymerization reaction of thermo-responsive films attached to the inner surface of PGMA nanotubes. PNIPAAm has been reported to switch between a hydrated, expanded state and a collapsed state across LCST.27 The mixture of the monomer NIPAAm with the photoinitiator AIBN and the crosslinker MBAA was vacuum-injected into the crosslinked PGMA nanotube backbones embedded in cylindrical alumina nanopores. Then the pendant PNIPAAm layer was formed inside the PGMA nanotubes via free-radical UV-polymerization. Finally the uniform long polymer nanotubes with crosslinked PGMA as the backbone and the pendant PNIPAAm layer inside as the thermo-responsive gating system were obtained following drying, mechanical polishing, the dissolution of the AAO template, dialysis to remove amorphous polymer impurities, centrifugation and cleaning.
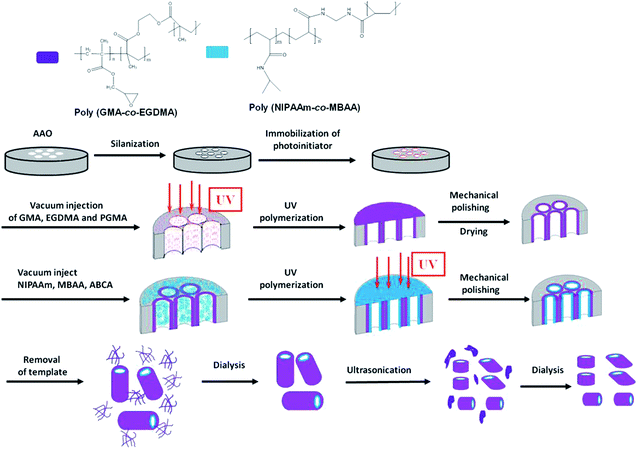 |
| | Fig. 1 Schematic of fabricating uniform long and fragmented PGMA-PNIPAAm nanotubes with the thermo-responsive gating system. | |
SEM and TEM images confirmed the success in fabricating the uniform long polymer nanotubes with crosslinked PGMA as the backbone and the pendant PNIPAAm layer inside as the thermo-responsive gating system as shown in Fig. 2. The outer diameter of the nanotubes is determined by the diameter of the pores in the AAO template while the length is determined by the thickness of the template, thus giving us an advantage in controlling the dimensions of the nanotubes. Even though the inner diameter is not easily controlled resulting in a tendency for the wall thickness to vary, the as-prepared nanotubes have shown the characteristics of uniformity as well as the flexibility to adjust on command. Furthermore, the amorphous polymer impurities accompanied with the synthesis of the nanotubes could be cleaned out by dialysis using a cellulose membrane in Nanopure water. There exists certain amount of amorphous polymer byproducts accompanied with the formation of PGMA-PNIPAAm NTs before dialysis (Fig. 2a). But a majority of such amorphous polymer impurities were dialyzed out from the PGMA-PNIPAAm NTs due to their relatively small molecular weight (Fig. 2b). Moreover, the SEM image in Fig. 2b clearly shows that the nanotubes have smooth outer surfaces and uniform dimensions with an outer diameter of 240 ± 6 nm, and a length of 60 ± 2 μm that resulted from introduction of the precursor materials consisting of 70% (v/v) GMA monomer, 30% (v/v) EGDMA crosslinker and 1% (w/v) pre-synthesized PGMA to the photo-initiator immobilized AAO templates with the nominal pore size of 200 nm and a thickness of 60 μm, followed by immobilization of the PNIPAAm layer inside. The incorporation of the pre-synthesized polymer into the monomer solution as the precursor material not only promotes the increase in the modulus of elasticity of the resulting polymer nanotubes, but also favors the decrease in polymerization shrinkage.
 |
| | Fig. 2 SEM images of uniform PGMA-PNIPAAm long nanotubes with the thermo-responsive gating system (a) before dialysis and (b) after dialysis; (c) PGMA-PNIPAAm fragmented short nanotubes with the thermo-responsive gating system, and (d) TEM image of PGMA-PNIPAAm nanotubes. | |
Fragmented and purified short polymer nanotubes with the thermo-responsive gating system from sonication-induced scission
A successful drug delivery system requires nanotubes under a certain size for effective cellular uptake during endocytosis. The long polymer nanotubes mentioned above are not suitable for application in drug delivery since the length is in the range of scores of micrometers, which is far beyond the size required for the effective cellular uptake via endocytosis. To reach the goal, here the sonication-induced scission method was employed to ‘cut’ the long nanotubes into short fragments. The mechanism of ultrasonication for nanotube scission is shown in Fig. 3. It is known that gas bubbles are capable of resonance vibration.24 The sonicator tip in the medium first generates acoustic waves. Bubbles of different sizes are generated, with the larger ones closer to the sonicator. This bubble distribution creates an effective density gradient and then drives the convective current to establish an optimal cutting zone for the nanotubes. Bubbles smaller than the resonance size will attract each other, then collide and coalesce through a “rectified diffusion” process. This is called nucleation. Once these bubbles gather and reach the critical resonance size, they start collapsing and create a strong hydrodynamic flow. This strong flow gradually pulls the nanotubes in the media to run parallel to the collapsing wall. A relative velocity between the nanotubes and the fluid is created and balanced by mechanical tensile stresses, which are maximal near the center of the nanotubes. After the stresses reach a critical level, nanotubes are pulled apart from the center of the nanotubes. The bubble then vanishes, leaving a cloud of small bubbles in the original space.21–23 We found that the sonication-induced scission only affects the length of the nanotubes, but not the diameter. No further length reduction was observed after 10 min sonication treatment. The data are not shown here. Fig. 1c shows the SEM image of the purified fragmented PGMA-PNIPAAm NTs with a length of 2 ± 0.5 μm, and an outer diameter of 240 ± 6 nm obtained after 10 min sonication treatment followed by dialysis using a cellulose membrane in Nanopure water. The more detailed TEM image (Fig. 2d) indicated the hollow structure of polymer nanotubes of 54 ± 4 nm in wall thickness and 130 nm in inner diameter. These purified fragmented short PGMA-PNIPAAm NTs provide us with an advantage for more efficient cellular internalization and drug loading compared to the original long nanotubes.
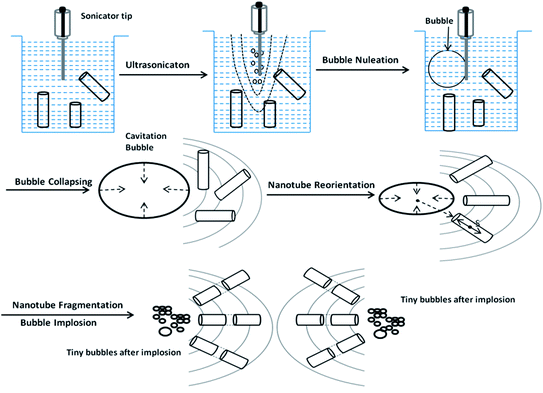 |
| | Fig. 3 Mechanism of sonication-induced scission for fragmentation of long polymer nanotubes. | |
Cytotoxicity of fragmented polymer nanotubes
The in vitro toxicity of thermo-responsive gating fragmented PGMA-PNIPAAm NTs with 5 wt% NIPAAm monomer was evaluated in KB-3-1 human cancer cells using MTT assays. The cells were incubated with polymer nanotubes for 72 h with varying concentrations from 0–250 mg L−1 at 25 and 37 °C respectively. The results shown in Fig. 4 revealed that the fragmented PGMA-PNIPAAm NTs have very low toxicity with the cell viability of 75–100% up to a tested concentration of 250 mg L−1 at both 25 and 37 °C. It indicated that pristine fragmented PGMA-PNIPAAm polymer nanotubes have good biocompatibility though toxic in their monomeric forms.
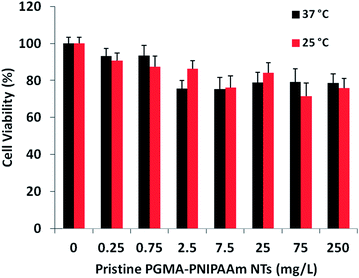 |
| | Fig. 4 Viability of KB-3-1 cancer cells incubated with pristine PGMA-PNIPAAM fragmented short nanotubes for 72 h at 25 and 37 °C. | |
The reversible thermo-responsive gating mechanism for drug loading and release from polymer nanotubes
One of the main objectives of targeted delivery is to achieve controlled release once the drug is at a specific location in the body. Stimuli-responsive gating is one way of achieving controlled release of the drugs on command. Here a typical thermo-responsive polymer, PNIPAAm, was used as the gating material to control drug release. PNIPAAm has a LCST of around 32 °C, close to the body temperature (37 °C). An increase in the LCST under physiological conditions would be desirable for biomedical applications including drug delivery, which could be achieved by co-polymerization with hydrophilic comonomers, such as acrylamide or N-isopropylmethacrylamide.28,29 However, as a proof of concept, no co-polymerization modification was performed on PNIPAAm to increase its LCST temperature in this report. The pendant crosslinked PNIPAAm layer attached to the inner surface of PGMA nanotubes underwent a discontinuous volume transition in response to a change in temperature of 32 °C at LCST, which can be attributed to the reversible formation and breakage of the hydrogen bonds between the water and hydrophilic groups of PNIPAAm and the hydrophilic/hydrophobic groups within their chains. Below LCST, a PNIPAAm hydrogel is hydrophilic and becomes “swollen” in aqueous media. In contrast, above the LCST, it undergoes a reversible phase transition and a sharp coil-to-globule transition to form inter- and intra-chain association, resulting in hydrophobic aggregation and volume collapse. Therefore, the ‘gates’ of nanotubes will be opened only when the temperature is above the LCST, otherwise they will be closed. Fig. 5 shows the drug loading, preservation, release and recovery for the PGMA-PNIPAAm nanotubes. Drugs can be loaded into nanotubes when their ‘gates’ are open in response to temperature changes. The nanotubes are completely immersed in a drug solution at a high temperature (here 37 °C) above LCST for a sufficiently long time to allow the drugs to diffuse to the opened nanotubes until equilibrium is reached. When the temperature (here 25 °C) is below LCST, the PNIPAAm layer inside the PGMA tubes swells and the ‘gates’ are in the closing state. Thus the drug will be held inside the nanotubes until thermal activation. The loaded drugs will be released from the nanotubes when the temperature of the surrounding media is increased since the PNIPAAm inner layer shrinks towards its original position leaving the gates of nanotubes open. Due to the hydrophobic interaction between the loaded drug and the hydrophobic shrunk PNIPAAm layer, the loaded drug will diffuse out of the nanotubes gradually, not abruptly.
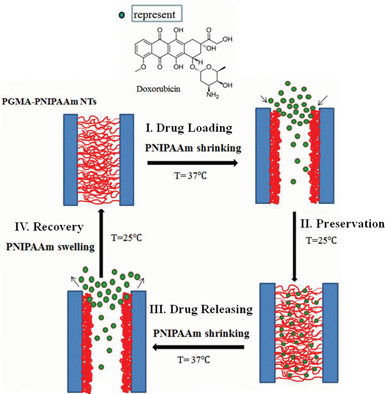 |
| | Fig. 5 Schematic of thermo-responsive loading/release behavior of DOX from the fragmented PGMA-PNIPAAm nanotubes. (I) Drug loading: the PGMA-PNIPAAm fragmented short nanotubes were loaded with DOX at a high temperature of 37 °C with the ‘gates’ opening; (II) preservation: the drugs are stored inside the polymer nanotubes at a low temperature of 25 °C with the ‘gates’ closing; (III) drug releasing: when the system is heated to 37 °C, the ‘gates’ of the nanotubes open releasing the loaded drugs; (IV) recovery: cooling nanotubes back to the initial shape. | |
Thermo-responsive release behavior of doxorubicin from the PGMA-PNIPAAm NTs and its kinetic studies
Doxorubicin is the preferred drug for model systems when they are tested in in vitro systems. The thermo-dependent DOX releasing from PGMA-PNIPAAm NTs with 5 wt% NIPAAm was investigated by fluorescence spectroscopy. In vitro DOX release profiles of the long and fragmented nanotubes are shown in Fig. 6a and b at two temperatures of 25 and 37 °C. At 25 °C, a negligible amount of DOX release was observed from both long and short PGMA-PNIPAAm NTs and the release percentage was kept almost at zero. As the temperature was increased to 37 °C, the amount of released DOX from long and fragmented nanotubes increased, but they represented different release behaviors in which the release rate of fragmented short nanotubes was slightly faster than long nanotubes due to the faster diffusion of DOX from the fragmented short nanotubes. The release trend lines of long and fragmented nanotubes crossed after the next 2.4 h, which gave around 36.0% DOX release percentage. After this point, the release rate of long nanotubes was higher than that of fragmented nanotubes. Finally, 67.47% and 43.66% DOX were released after 11 h for long and fragmented PGMA-PNIPAAm NTs respectively.
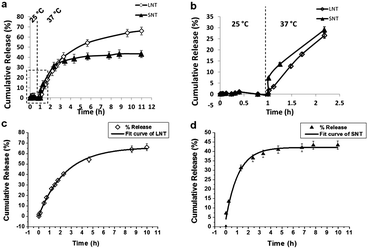 |
| | Fig. 6 (a) Thermo-responsive release behaviour of DOX from long PGMA-PNIPAAm nanotubes (LNTs) and fragmented short PGMA-PNIPAAm (SNTs); (b) zoom part for DOX release behavior upon temperature change; and (c) the first order kinetic model fitting curve of DOX release behavior for LNTs; (d) the first order kinetic model fitting curve of DOX release behavior for SNTs. | |
Although the mechanism of drug release from polymeric matrices is very complex and is still not completely understood, it can be simplistically classified as either pure diffusion, erosion controlled release or a combination of the two mechanisms.30 Model dependent methods based on different mathematical functions can be used to determine the suitable drug release kinetic model. The drug release following the first order kinetics can be expressed by the equation:
| | | y(t) = y0(1 − exp(−kt)) | (1) |
where
y(
t) is the accumulative drug release percentage at time
t,
k is the first order rate constant expressed in units of inverse of time, and
y0 is the final accumulative drug release percentage. The DOX release percentage
y(
t) as a function of time varied between PGMA-PNIPAAm long nanotubes and fragmented short nanotubes (
Fig. 6b and c). The curves resulting from regression analysis of the data by
eqn (1) from the first order kinetic relationship were obtained for long nanotubes and fragmented nanotubes respectively as follows:
| | | y(t) = 65.8(1 − e−0.42t) | (2) |
| | | y(t) = 41.7(1 − e−1.09t) | (3) |
65.8% and 41.7% are the final theoretical DOX release percentage values after 11 h for long polymer nanotubes and fragmented short polymer nanotubes, respectively, which are very close to the experimental magnitude of 67.47% and 43.66%. Visual inspection reveals that
eqn (2) and
(3) describe the release data of
Fig. 6b and c very well. The result demonstrates that the DOX release rate in fragmented short nanotubes is slower than long nanotubes, which means short polymer nanotubes have a stronger hydrophobic effect towards DOX drug molecules.
In vitro investigation of DOX-loaded polymer nanotubes on the cell viability
Nanotubes can be internalized by cancer cells and may provide a vehicle with which to deliver chemotherapeutic agents to the interior of cells, causing apoptosis.5,11 To investigate the fragmented short PGMA-PNIPAAm NTs as a viable controllable delivery strategy, we tested the delivery of the chemotherapeutic drug DOX to KB-3-1 human epidermoid carcinoma cells. The in vitro cytotoxicity of DOX-loaded fragmented short PGMA-PNIPAAm NTs against KB-3-1 cells determined by the MTT assay is shown in Fig. 7(a). We observed thermo-responsive cytotoxicity in short polymer nanotubes. At 25 °C, 53.72% cell viability was obtained for DOX-loaded fragmented nanotubes at a DOX dosage of 10 μmol L−1. However, cell viabilities decreased to about 11.12% for DOX-loaded short polymer nanotubes when the temperature was increased to 37 °C under otherwise the same conditions. Furthermore, PGMA-PNIPAAm NTs containing DOX exhibit concentration-dependent toxicity at 37 °C. The observed drug dose required for 50% cellular growth inhibition (IC50) of DOX-loaded PGMA-PNIPAAm NTs is 1.40 μmol L−1, which was a little higher than that for free DOX (IC50 = 0.3 μmol L−1). However, significant reduced cell viabilities to about 37–40% and 9–12% were observed at a DOX dosage of 3 and 10 μmol L−1 respectively. When the cell viability of DOX-loaded SNTs is divided by that of free DOX at the corresponding concentrations, we observe a drastic difference in cell viability at 25 °C with the highest DOX concentration as shown in Fig. 7(b). As the concentration of DOX increases from 0.01 to 10 μmol L−1, the cell survival ratio significantly increases at 25 °C. On the other hand, the cell survival rate for DOX-loaded SNTs at 37 °C is almost similar to the control. At 10 μmol L−1 of DOX concentration, the cell survival rate for the DOX-loaded SNTs at 25 °C is 12 times that for the control while the rate for the DOX-loaded SNTs at 37 °C is less than 3 times that for the control. This means DOX is successfully preserved inside the nanotubes at 25 °C while successful drug release was achieved at 37 °C. As compared to other delivery systems reported,31–33 such a good thermo-responsive cytotoxicity with low IC50 shows the superiority of PGMA-PNIPAAm fragmented nanotubes developed in this work. Thus, the novel polymer nanotubes may serve as effective intracellular thermo-triggered drug delivery systems.
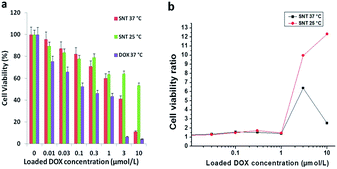 |
| | Fig. 7 (a) Viability of KB-3-1 cancer cells incubated with DOX-loaded PGMA-PNIPAAM SNTs for 72 h at 25 °C and 37 °C, and free DOX respectively; (b) the ratios of cell viability at 25 °C and 37 °C for DOX-loaded SNTs respectively to that for free DOX at the corresponding concentrations. | |
Conclusions
A simple and effective method has been developed to synthesize novel polymer nanotubes with a thermo-responsive gating system. A sonication-induced scission method was employed to ‘cut’ long nanotubes into fragmented short ones, which could be used as superior nanocarriers for efficient delivery and release of anti-cancer drugs into cancer cells. The ‘gates’ of nanotubes can be reversibly controlled between ‘closing’ and ‘opening’ states upon a temperature change across the LCST. The results obtained from this work demonstrated that PGMA-PNIPAAm NTs showed great promise of therapeutic efficacy for potential anticancer treatment. Further investigations on the developed anti-cancer drug delivery systems with higher LCST under physiological conditions (37 °C) with multi-drug resistant cancer cells and in vivo studies remain underway. And intracellular drug release behaviours of the drug-loaded nanotubes will be real-time monitored as well. With the unique structural characteristics of polymer nanotubes one can build up multifunctional drug delivery systems including imaging, targetability and therapy for the diagnosis and therapeutic treatment of cancer disease.
Acknowledgements
We acknowledge St. John's University for the Seed Grant support. And we thank Lihua Zhang from Brookhaven National Laboratory for the help with TEM imaging. Research was carried out in part at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of energy, Office of Basic Energy Sciences, under Contract no. DE-AC02-CH10886.
References
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- P. V. Devarajan, A. B. Jindal, R. R. Patil, F. Mulla, R. V. Gaikwad and A. Samad, J. Pharm. Sci., 2010, 99, 2576–2581 CrossRef CAS PubMed.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. R. Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef CAS PubMed.
- Y. Lee, G.-E. Jung, S. J. Cho, K. E. Geckeler and H. Fuchs, Nanoscale, 2013, 5, 8577–8585 RSC.
- A. Nan, X. Bai, S. J. Son, S. B. Lee and H. Ghandehari, Nano Lett., 2008, 8, 2150–2154 CrossRef CAS PubMed.
- G. Chen, Pharm. Anal. Acta, 2012, 3, 1000e1136 Search PubMed.
- S. Kapoor and A. J. Bhattacharyya, J. Phys. Chem. C, 2009, 113, 7155–7163 CAS.
- Y.-J. Yang, X. Tao, Q. Hou, Y. Ma, X.-L. Chen and J.-F. Chen, Acta Biomater., 2010, 6, 3092–3100 CrossRef CAS PubMed.
- S. J. Son, J. Reichel, B. He, M. Schuchman and S. B. Lee, J. Am. Chem. Soc., 2005, 127, 7316–7317 CrossRef CAS PubMed.
- M. R. Abidian, D.-H. Kim and D. C. Martin, Adv. Mater., 2006, 18, 405–409 CrossRef CAS PubMed.
- N. W. S. Kam and H. Dai, J. Am. Chem. Soc., 2005, 127, 6021–6026 CrossRef CAS PubMed.
- J. Grumelard, A. Taubert and W. Meier, Chem. Commun., 2004, 1462–1463 RSC.
- X. Ding, N. Kameta and M. Masuda, Pharm Tech Jpn., 2013, 29, 2313–2322 CAS.
- E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, P. Amoros and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS PubMed.
- R. Liu, X. Zhao, T. Wu and P. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- C.-L. Zhu, C.-H. Lu, X.-Y. Song, H.-H. Yang and X.-R. Wang, J. Am. Chem. Soc., 2011, 133, 1278–1281 CrossRef CAS PubMed.
- X.-U. Jiang, C. Roecker, M. Hafner, S. Brandholt, R. M. Doerlich and G. U. Nienhaus, ACS Nano, 2010, 4, 6787–6797 CrossRef CAS PubMed.
- M. S. Cartiera, K. M. Johnson, V. Rajendran, M. J. Caplan and W. M. Saltzman, Biomaterials, 2009, 30, 2790–2798 CrossRef CAS PubMed.
- S.-D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496–504 CrossRef CAS PubMed.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- F. Hennrich, R. Krupke, K. Arnold, J. A. Rojas Stuetz, S. Lebedkin, T. Koch, T. Schimmel and M. M. Kappes, J. Phys. Chem. B, 2007, 111, 1932–1937 CrossRef CAS PubMed.
- Y. Y. Huang, T. P. J. Knowles and E. M. Terentjev, Adv.
Mater., 2009, 21, 3945–3948 CrossRef CAS.
- A. Lucas, C. Zakri, M. Maugey, M. Pasquali, P. van der Schoot and P. Poulin, J. Phys. Chem. C, 2009, 113, 20599–20605 CAS.
- D. E. Hughes and W. L. Nyborg, Science, 1962, 138, 108–114 CAS.
- C. Zou, Z. Luo, D. H. Le, K. Dessources, A. Robles and G. Chen, J. Mater. Chem., 2011, 21, 14543–14548 RSC.
- G. Chen, S. A. Soper and R. L. McCarley, Langmuir, 2007, 23, 11777–11781 CrossRef CAS PubMed.
- G. Joseph, J. Pichardo and G. Chen, Analyst, 2010, 135, 2303–2308 RSC.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- G. Dalkas, K. Pagonis and G. Bokias, Polymer, 2006, 47, 243–248 CrossRef CAS PubMed.
- C. R. Young, C. Dietzsch, M. Cerea, T. Farrell, K. A. Fegely, A. Rajabi-Siahboomi and J. W. McGinity, Int. J. Pharm., 2005, 301, 112–120 CrossRef CAS PubMed.
- R. Cheng, X. Wang, W. Chen, F. Meng, C. Deng, H. Liu and Z. Zhong, J. Mater. Chem., 2012, 22, 11730–11738 RSC.
- D. Sutton, N. Nasongkla, E. Blanco and J. Gao, Pharm. Res., 2007, 24, 1029–1046 CrossRef CAS PubMed.
- L. Zhou, R. Cheng, H. Tao, S. Ma, W. Guo, F. Meng, H. Liu, Z. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 1460–1467 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 





